

جلد 23، شماره 1 - ( بهار 1405 )
جلد 23 شماره 1 صفحات 77-61 |
برگشت به فهرست نسخه ها

Ethics code: IR.TMI.RCE.1398.014
![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Roshanzamir F, Mohajer Ansari J, Norouzian M, Teremmahi Ardestani M. Therapeutic approches for Graft-versus-Host Disease Risk Reduction in Mouse Models of Hematopoietic Stem Cells Transplantation. bloodj 2026; 23 (1) :61-77
URL: http://bloodjournal.ir/article-1-1611-fa.html
URL: http://bloodjournal.ir/article-1-1611-fa.html
روشن ضمیر فاطمه، مهاجر انصاری جواد، نوروزیان مرضیه، ترماحی اردستانی* مجید. راهکارهای درمانی کاهش خطر بیماری پیوند علیه میزبان در مدل موشی با پیوند سلولهای بنیادی خونساز. فصلنامه پژوهشی خون. 1405; 23 (1) :61-77



مرکز تحقیقات پزشکی مولکولی پژوهشکده سلامت هرمزگان & مرکز تحقیقات غذذ و متابولیسم دانشگاه علوم پزشکی هرمزگان
متن کامل [PDF 1122 kb]
(92 دریافت)
| چکیده (HTML) (180 مشاهده)


1) مهار فعالسازی سلولهای T مانند آنتیبادی ضد CD28 و CD26 که با کاهش هم تحریکی و پاسخ التهابی، GvHD را محدود میکنند. 2) مهار مهاجرت سلولی مانند CCR5 و FTY720 که بر مهاجرت سلولهای T اثر میگذارد؛ هر چند نتایج برای CCR5 مشابه و قانعکننده نبوده است. 3) مداخلات سلولی و ایمنی مانند CAR-T و CD56+CD3+ که بیشتر بر تعادل میان اثر GVL و کنترل GvHD تمرکز دارند. مقایسه مکانیسم اثر، مزایا و محدودیتها، تأثیر بر GvHD و GVL مداخلات ذکر شده در مطالعه در جدول 3 ارائه شده است. مطالعههای مختلف نشان دادهاند که هدف قرار دادن مسیرهای کلیدی فعالسازی سلولهای T، مهار سیتوکینهای التهابی و کنترل عملکرد سلولهای APC هم شدت GvHD را کاهش دادهاند و هم اثر GVL را حفظ کردهاند؛ حتی در برخی موارد GVL تقویت شده است (42، 38، 37، 24، 17، 13، 11).
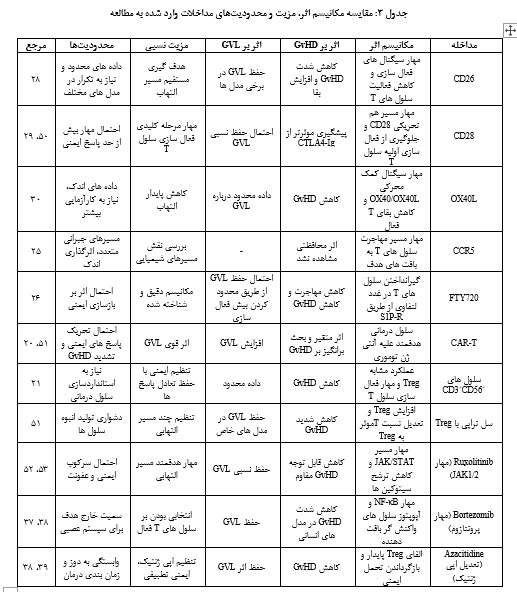 همچنین درمان های سلولی نشان دادهاند که تقویت جمعیت سلولهای T تنظیمی هم از طریق تزریق مستقیم و هم از طریق القای آنها با آنتیبادیها یا مولکولهای خاص مانند anti-CD137 از مؤثرترین راهبردها برای مهار GvHD همراه با حفظ اثر GVL است (44، 43، 35، 33). به طور کلی نتایج نشان دادهاند که تمرکز بر مهار مسیرهای اختصاصی فعالسازی سلولهای T، القای تحمل ایمنی و استفاده از سلولهای تنظیمی یا NK فعال شده کارآمدترین رویکردها در ایجاد تفکیک عملکردی میان GvHD و GVL بودهاند. در مقابل روشهای سرکوب ایمنی غیر اختصاصی و گسترده، هر چند موجب کاهش GvHD میشوند اما معمولاً با کاهش GVL همراه هستند (9). در میان رویکردهای درمانی، مهار مسیرهای فعالسازی و مهاجرت سلولهای T نقش مهمی در کنترل GvHD دارد. برای مثال، گزارش شده است که مهارکننده های CCR5 اثر محافظتی قابل توجهی در کاهش شدت GvHD نشان ندادهاند (25). در مقابل، فینگولیمود (FTY720) با مهار مهاجرت سلولهای T از غدد لنفاوی در مدلهای موشی موجب کاهش GvHD میشود (26). هم چنین مهارکنندههای CD26 نیز به عنوان یکی از اهداف بالقوه در کاهش GvHD معرفی شدهاند (28). علاوه بر این، مهار OX40L در مدلهای موشی با کاهش GvHD همراه بوده و در عین حال فعالیت GVL را حفظ کرده است (30).
همچنین درمان های سلولی نشان دادهاند که تقویت جمعیت سلولهای T تنظیمی هم از طریق تزریق مستقیم و هم از طریق القای آنها با آنتیبادیها یا مولکولهای خاص مانند anti-CD137 از مؤثرترین راهبردها برای مهار GvHD همراه با حفظ اثر GVL است (44، 43، 35، 33). به طور کلی نتایج نشان دادهاند که تمرکز بر مهار مسیرهای اختصاصی فعالسازی سلولهای T، القای تحمل ایمنی و استفاده از سلولهای تنظیمی یا NK فعال شده کارآمدترین رویکردها در ایجاد تفکیک عملکردی میان GvHD و GVL بودهاند. در مقابل روشهای سرکوب ایمنی غیر اختصاصی و گسترده، هر چند موجب کاهش GvHD میشوند اما معمولاً با کاهش GVL همراه هستند (9). در میان رویکردهای درمانی، مهار مسیرهای فعالسازی و مهاجرت سلولهای T نقش مهمی در کنترل GvHD دارد. برای مثال، گزارش شده است که مهارکننده های CCR5 اثر محافظتی قابل توجهی در کاهش شدت GvHD نشان ندادهاند (25). در مقابل، فینگولیمود (FTY720) با مهار مهاجرت سلولهای T از غدد لنفاوی در مدلهای موشی موجب کاهش GvHD میشود (26). هم چنین مهارکنندههای CD26 نیز به عنوان یکی از اهداف بالقوه در کاهش GvHD معرفی شدهاند (28). علاوه بر این، مهار OX40L در مدلهای موشی با کاهش GvHD همراه بوده و در عین حال فعالیت GVL را حفظ کرده است (30).
در حوزه درمانهای سلولی نیز نتایج قابل توجهی گزارش شده است. در یک مدل موش، تزریق CAR آلوژنیک CD19 با کمک محرک CD28 موجب افزایش فعالیت GVL همراه با حداقل GvHD شده است، هر چند تأثیر دقیق آن بر GvHD همچنان مورد بحث است (50). همچنین گزارش شده است که سلولهایی با زیر جمعیت CD56+CD3+ میتوانند عملکردی مشابه Treg داشته باشند و از طریق مهار پاسخهای ایمنی، موجب کاهش GvHD شوند (51). در سالهای اخیر، رویکردهای نوین درمانی با تمرکز بر سلول درمانی، به ویژه سلولهای MSC و نیز هدفگیری مسیرهای سیگنالینگ مانند JAK/STAT با استفاده از مهارکنندههایی نظیر ruxolitinib ، نتایج امیدوارکنندهای در کاهش شدت GvHD و حفظ اثر GVT نشان دادهاند (54-52). علاوه بر مداخلاتی که توانستهاند در کاهش شدت GVHD مؤثر باشند، برخی مطالعهها نتایج منفی یا عوارض جانبی نشان دادهاند. به عنوان مثال، مهار مسیر CCR5 در برخی مدلها نتوانست اثر محافظتی قابلتوجهی در کاهش GvHD ایجاد کند که این موضوع میتواند به وجود مسیرهای جایگزین در مهاجرت و فعالسازی سلولهای T نسبت داده شود. این یافته نشان میدهد که مهار یک مسیر منفرد ممکن است برای کنترل کامل پاسخ التهابی کافی نباشد (25). از سوی دیگر، برخی روشهای درمانی مانند CAR‑T اگرچه اثر GVL قابلتوجهی دارند، اما در مورد تأثیر آنها بر GvHD نتایج یکنواختی گزارش نشده و در برخی موارد احتمال تشدید پاسخ ایمنی و افزایش التهاب سیستمیک وجود دارد (50). همچنین، مداخلاتی که مسیرهای کو محرکهای سلول T مانند CD28 را هدف قرار میدهند، در صورت مهار بیش از حد، ممکن است به کاهش شدید پاسخ ایمنی و ایجاد اختلال در تعادل ایمنی منجر شوند.
یکی از محدودیتهای تفسیر جامع نتایج مطالعههای آزمایشگاهی در مدلهای موشی، ناهمگونی مدلهای موشی پیوند است که میتواند به طور مستقیم بر شدت GvHD و پاسخ به مداخلات درمانی اثر بگذارد. تفاوتهای ژنتیکی میان دهنده و گیرنده، درجات متفاوتی از فعالسازی سلولهای T و در نتیجه شدتهای مختلف GvHD ایجاد میکنند. به علاوه، نوع نقص ایمنی گیرنده نیز عامل تعیین کنندهای است. مدلهای مبتنی بر اشعه با آسیب مخاطی، التهاب شدیدتری ایجاد میکنند؛ در حالی که مدلهای دارویی یا موشهای انسانی شده مانند NSG الگوی متفاوت و اغلب شدیدتری از GvHD نشان میدهند. این تفاوتها باعث تغییر اثربخشی مداخلات انجام شده میگردد. به طور کلی، این تفاوتها نشان میدهند که تفسیر نتایج درمانی باید با توجه به ویژگیهای ژنتیکی و ایمنی مدل استفاده شده انجام شود و تعمیم نتایج به سایر مدلها و انسان باید با احتیاط صورت گیرد.
نتیجهگیری
با توجه به بروز بالای عوارض و مرگومیر ناشی از GvHD، درک مکانیسمهای اصلی درگیر در این عارضه برای شناسایی راهکارهای درمانی مؤثر اهمیت دارد. در اغلب مدلهای آزمایشگاهی، ابتدا سلولهای PBMC انسانی به موشهای تحت تابش تزریق شده و سپس مداخلات مختلف برای کاهش GvHD ارزیابی شدهاند (38، 37). یکی از چالشهای اصلی، ایجاد تعادل میان مهار GvHD و حفظ اثر ضد توموری پیوند است؛ به طوری که برخی مداخلات مانند آزاتیدین و مهارکنندههای پروتئازوم توانستهاند هر دو هدف را همزمان حفظ کنند (38، 37). این تعادل تحت تأثیر نوع مداخله، زمان اعمال آن و نقش زیر جمعیتهای سلولهای T و سلولهای ایمنی ذاتی مانند NK و MDSC قرار دارد (38، 37). در برخی راهبردها مانند واکسیناسیون علیه WT1، افزایش اثر GVT همراه با کنترل GvHD مشاهده شده است (55، 49). همچنین، مدلهای انسانیشده مانند NSG، NOG و BRG ابزارهای مهمی برای مطالعه مکانیسمها و ارزیابی درمانهای نوظهور محسوب میشوند. شواهد نشان میدهد که مداخلات هدفمند علیه مسیرهای فعالسازی و مهاجرت سلولهای T بیشترین ظرفیت را برای کاهش شدت GvHD دارند، اگرچه اثر آنها بر حفظ GVL در مطالعههای مختلف متفاوت گزارش شده است. با وجود پیشرفتها، ناهمگونی در طراحی مطالعهها و محدودیتهای مدلهای حیوانی موش، تعمیمپذیری نتایج را کاهش میدهد. بهبود طراحی مدلها، افزایش استانداردسازی، بررسی ترکیب درمانها و توجه به مطالعههایی که نتایج منفی یا عوارض ناخواسته نشان دادهاند، میتواند به درک محدودیتهای هر روش و ارائه راهبردهای دقیقتر و مؤثرتر برای کنترل پایدار GvHD کمک کند (38، 37).
حمایت مالی
این پژوهش با حمایت معنوی دانشگاه علوم پزشکی بندرعباس انجام شده است.
نقش نویسندگان
دکتر فاطمه روشن ضمیر: نگارش نسخه نهایی و ویرایش نهایی مقاله
دکتر جواد مهاجر انصاری: ویرایش مقاله
دکتر مرضیه نوروزیان: نگارش مقاله
دکتر مجید ترماحی اردستانی: جمعآوری مطالب و نگارش نسخه اولیه مقاله
متن کامل: (27 مشاهده)
راهکارهای درمانی کاهش خطر بیماری پیوند علیه میزبان در مدل موشی با
پیوند سلولهای بنیادی خونساز
فاطمه روشنضمیر1، 2 ، جواد مهاجر انصاری2 ، مرضیه نوروزیان2 ، مجید ترماحی اردستانی3
1- مرکز تحقیقات پزشکی مولکولی پژوهشکده سلامت هرمزگان ـ دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
2- مرکز تحقیقات غدد و متابولیسم ـ دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
3- گروه علوم آزمایشگاهی ـ دانشکده پیراپزشکی دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
پیوند سلولهای بنیادی خونساز
1- مرکز تحقیقات پزشکی مولکولی پژوهشکده سلامت هرمزگان ـ دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
2- مرکز تحقیقات غدد و متابولیسم ـ دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
3- گروه علوم آزمایشگاهی ـ دانشکده پیراپزشکی دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران
  http://dx.doi.org/10.61186/bloodj.22.1.11 Citation: Roshanzamir F, Mohajer Ansari j, Norouzian M, Teremmahi Ardestani M. Therapeutic approches for Graft-versus-Host Disease Risk Reduction in Mouse Models of Hematopoietic Stem Cells Transplantation. J Iran Blood Transfus. 2026: 23 (1): 61-77 نویسنده مسئول: دکتر فاطمه روشن ضمیر. استادیار مرکز تحقیقات پزشکی مولکولی پژوهشکده سلامت هرمزگان و مرکز تحقیقات غدد و متابولیسم ـ دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران کد پستی: 79166-13885 E-mail: fatemelab@gmail.com
نویسنده مسئول: دکتر مجید ترماحی اردستانی. استادیار گروه علوم آزمایشگاهی، دانشکده پیراپزشکی، دانشگاه علوم پزشکی هرمزگان ـ بندرعباس ـ ایران کد پستی: 79166-13885 E-mail: majidardestani50@gmail.com |
چکیده سابقه و هدف بیماری پیوند علیه میزبان (GvHD) ، یکی از مهمترین و جدیترین عوارض پس از پیوند سلولهای بنیادی خونساز (HSCT) است که میتواند به طور قابل توجهی بر بقای بیمار، کیفیت زندگی و موفقیت درمان تأثیر بگذارد. این مطالعه با هدف مرور استراتژیهای کاهش GvHD در مدلهای موشی و با تأکید بر نقش سلولهای بنیادی خونساز و سازوکارهای ایمنی مؤثر در این فرایند انجام شد. مواد و روشها این مطالعه مروری با جستجو در پایگاههای PubMed، Scopus و Web of Science طی سالهای ۲۰۰۰ تا ۲۰۲5 انجام شد. از میان ۱۲۰۰ مطالعه اولیه، پس از غربالگری، ۱۵۵ مقاله واجد معیارهای ورود شناسایی شدند و در نهایت ۵5 مطالعه برای تحلیل نهایی مورد بررسی قرار گرفتند. مطالعات منتخب به طور عمده شامل مداخلات دارویی، مونوکلونال آنتیبادیها، مهارکنندههای مسیرهای سیگنالینگ و درمانهای سلولی بودند. یافتهها نتایج نشان داد که راهبردهای دارویی مانند سیکلوسپورین و آزاسیتیدین، آنتیبادیهای مونوکلونال مانند توسیلیزوماب، مهارکنندههای مسیرهای سیگنالینگ بهویژه JAK/STAT و روشهای سلولی شامل Tregو MSC، بهطور معناداری موجب کاهش بروز و شدت GvHD در مدلهای موشی شدند. همچنین، مطالعههای اخیر اثربخشی سلول های MSCرا در درمان بیماریهای مختلف و نقش مهارکنندههای JAK را در کنترل GvHD تأیید کردهاند. در بسیاری از این مطالعهها، اثر ضد توموری پیوند (GVT) نیز تا حد زیادی حفظ شد که نشاندهنده اهمیت انتخاب درمانهای هدفمند و متعادل است. نتیجه گیری این مطالعه نشان داد که مدلهای موشی دچار نقص ایمنی، ابزارهایی ارزشمند برای بررسی پاتوفیزیولوژی GvHD و ارزیابی درمانهای نوین محسوب میشوند. استفاده از راهبردهای نوین ایمونوتراپی، تنظیم پاسخ های التهابی و بهره گیری از سلول های تنظیمی می تواند نقش موثری در کاهش شدت GvHD داشته باشد. بهکارگیری رویکردهای ترکیبی، انتخاب مداخله متناسب با شرایط بیمار و شخصیسازی درمان میتواند به ایجاد تعادل بین کنترل GvHD و حفظ اثر ضد توموری کمک کند. با این حال تفاوت میان مدل های موشی و شرایط بالینی انسانی همچنان یکی از موانع اصلی انتقال این یافته ها به درمان است. بنابراین انجام مطالعات بیشتر برای تعیین ایمنی، اثریخشی و بهینه سازی زمان و دوز درمان ها ضروری به نظر می رسد. کلمات کلیدی: بیماری پیوند علیه میزبان، سیتوکینها، ایمونوتراپی، کموکینها، درمان |
مقدمه
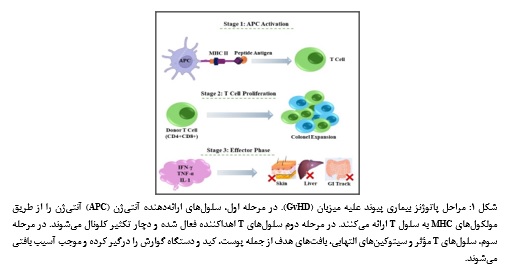
پیوند سلولهای بنیادی خونساز (HSCT : Hematopoietic Stem Cell Transplantation) یکی از مهمترین روشهای درمانی برای بسیاری از بدخیمیهای خونی و اختلالات هماتولوژیک محسوب میشود. با وجود اثربخشی بالینی این روش، بروز بیماری پیوند علیه میزبان (GvHD : Graft versus Host Disease) همچنان یکی از مهمترین محدودیتهای آن است و مرگ و میر بالایی دارد. این عارضه ناشی از حمله لنفوسیتهای Tاهداکننده به آنتیژنهای بافت گیرنده (میزبان) است. در مدلهای انسانی و حیوانی، این بیماری به دو شکل حاد و مزمن مشاهده میشود. در شکل حاد، علائم در کمتر از ۳ ماه پس از پیوند ظاهر میشوند. معمولاً پوست، دستگاه گوارش و کبد به شدت درگیر شده و بثورات پوستی سطح بدن را میپوشانند. در مقابل، در شکل مزمن که پس از ۱۰۰ روز خود را نشان میدهد، معمولاً با درگیری مفاصل و سطوح مخاطی همراه است، جایی که علائم کاملاً شبیه به بیماران خود ایمنی است. به طور خلاصه مکانیسم بیماری داری سه مرحله است شامل فعالسازی سلولهای ارائه دهنده آنتیژن (Antigen Processing Cells, APCs)، فعالسازی، تکثیر، تمایز و مهاجرت سلولهای T و مرحله اجرایی که در این مرحله لنفوسیتهای T و سیتوکینها باعث تخریب و آسیب بافتی در اندامهای هدف میشوند (2، 1)(شکل 1). اگر چه در دهههای اخیر درک ما از پاتوفیزیولوژی GvHD، از جمله نقش فعالسازی سلولهای T، سیتوکینها، مسیرهای سیگنالینگ و تعاملات سلولهای ایمنی ذاتی و اکتسابی، به طور چشمگیری گسترش یافته است، اما کنترل بالینی این بیماری همچنان با چالش های فراوان همراه است (3، 1). در حال حاضر، کورتیکواستروئیدها درمان خط اول GvHD حاد محسوب میشوند، با این حال پاسخ به آنها در بخش قابل توجهی از بیماران ناکافی بوده و مقاومت یا عود بیماری، نیاز به درمانهای خط دوم و راهبردهای هدفمندتر را برجسته میکند. در این راستا، رویکردهای متعددی شامل مهار سیتوکینها، آنتیبادیهای مونوکلونال، تعدیل مسیرهای سیگنالینگ داخل سلولی، افزایش سلولهای T تنظیمی (Regulatory T Cell, Treg)، سلولهای NK (Natural Killer Cell)، MSC (Mesenchymal Stem Cell) و سایر درمانهای ایمونومدولاتوری مورد بررسی قرار گرفتهاند؛ با این حال، بسیاری از این راهبردها هنوز با محدودیتهایی از نظر اثربخشی و ایمنی مواجه هستند. از سوی دیگر، مدلهای حیوانی، به ویژه مدلهای موشی، نقش مهمی در روشنسازی مکانیسمهای بیماری و ارزیابی درمانهای جدید قبل از ورود به بالین ایفا کردهاند. با وجود پیشرفتهای قابل توجه، هنوز یک شکاف مهم میان یافتههای آزمایشگاهی و کاربرد موفق بالینی وجود دارد و نیاز به بررسی و جمع بندی راهبردهای مؤثر همچنان احساس میشود (11-4). بر این اساس، مطالعه حاضر با هدف مرور و طبقهبندی راهبردهای کاهش GvHD در مدلهای موشی، با تمرکز بر مداخلات ایمونوتراپی، مسیرهای مولکولی هدف و عوامل تعدیلکننده پاسخ ایمنی، انجام شد تا تصویری جامع از وضعیت دانش موجود و جهتگیریهای آینده این حوزه ارائه دهد.
پیوند سلولهای بنیادی خونساز (HSCT : Hematopoietic Stem Cell Transplantation) یکی از مهمترین روشهای درمانی برای بسیاری از بدخیمیهای خونی و اختلالات هماتولوژیک محسوب میشود. با وجود اثربخشی بالینی این روش، بروز بیماری پیوند علیه میزبان (GvHD : Graft versus Host Disease) همچنان یکی از مهمترین محدودیتهای آن است و مرگ و میر بالایی دارد. این عارضه ناشی از حمله لنفوسیتهای Tاهداکننده به آنتیژنهای بافت گیرنده (میزبان) است. در مدلهای انسانی و حیوانی، این بیماری به دو شکل حاد و مزمن مشاهده میشود. در شکل حاد، علائم در کمتر از ۳ ماه پس از پیوند ظاهر میشوند. معمولاً پوست، دستگاه گوارش و کبد به شدت درگیر شده و بثورات پوستی سطح بدن را میپوشانند. در مقابل، در شکل مزمن که پس از ۱۰۰ روز خود را نشان میدهد، معمولاً با درگیری مفاصل و سطوح مخاطی همراه است، جایی که علائم کاملاً شبیه به بیماران خود ایمنی است. به طور خلاصه مکانیسم بیماری داری سه مرحله است شامل فعالسازی سلولهای ارائه دهنده آنتیژن (Antigen Processing Cells, APCs)، فعالسازی، تکثیر، تمایز و مهاجرت سلولهای T و مرحله اجرایی که در این مرحله لنفوسیتهای T و سیتوکینها باعث تخریب و آسیب بافتی در اندامهای هدف میشوند (2، 1)(شکل 1). اگر چه در دهههای اخیر درک ما از پاتوفیزیولوژی GvHD، از جمله نقش فعالسازی سلولهای T، سیتوکینها، مسیرهای سیگنالینگ و تعاملات سلولهای ایمنی ذاتی و اکتسابی، به طور چشمگیری گسترش یافته است، اما کنترل بالینی این بیماری همچنان با چالش های فراوان همراه است (3، 1). در حال حاضر، کورتیکواستروئیدها درمان خط اول GvHD حاد محسوب میشوند، با این حال پاسخ به آنها در بخش قابل توجهی از بیماران ناکافی بوده و مقاومت یا عود بیماری، نیاز به درمانهای خط دوم و راهبردهای هدفمندتر را برجسته میکند. در این راستا، رویکردهای متعددی شامل مهار سیتوکینها، آنتیبادیهای مونوکلونال، تعدیل مسیرهای سیگنالینگ داخل سلولی، افزایش سلولهای T تنظیمی (Regulatory T Cell, Treg)، سلولهای NK (Natural Killer Cell)، MSC (Mesenchymal Stem Cell) و سایر درمانهای ایمونومدولاتوری مورد بررسی قرار گرفتهاند؛ با این حال، بسیاری از این راهبردها هنوز با محدودیتهایی از نظر اثربخشی و ایمنی مواجه هستند. از سوی دیگر، مدلهای حیوانی، به ویژه مدلهای موشی، نقش مهمی در روشنسازی مکانیسمهای بیماری و ارزیابی درمانهای جدید قبل از ورود به بالین ایفا کردهاند. با وجود پیشرفتهای قابل توجه، هنوز یک شکاف مهم میان یافتههای آزمایشگاهی و کاربرد موفق بالینی وجود دارد و نیاز به بررسی و جمع بندی راهبردهای مؤثر همچنان احساس میشود (11-4). بر این اساس، مطالعه حاضر با هدف مرور و طبقهبندی راهبردهای کاهش GvHD در مدلهای موشی، با تمرکز بر مداخلات ایمونوتراپی، مسیرهای مولکولی هدف و عوامل تعدیلکننده پاسخ ایمنی، انجام شد تا تصویری جامع از وضعیت دانش موجود و جهتگیریهای آینده این حوزه ارائه دهد.
مواد و روشها
جهت انجام این مطالعه مروری، جست و جوی منابع در پایگاههای PubMed ،Scopus و Web of Science با استفاده از کلید واژههای مرتبط با GvHD، مدلهای حیوانی موش، مهارکنندههای سیتوکینـی، آنتــیبادیهای مــونوکلونال و مسیرهای سیگنالینگ، در بازه زمانی2000 تا 2025 انجام گرفت. معیارهای ورود شامل مطالعههای انگلیسی زبان دارای متن کامل بود که به بررسی مکانیسمهای ایجاد یا راهکارهای کاهش GvHD در مدلهای حیوانی، به ویژه موش، پرداخته بودند. مقالاتی که صرفاً بر مدل انسانی یا کشت سلولی متمرکز بودند، مقالات مروری و متاآنالیزها، و گزارشهای ناقص از مطالعه حذف شدند. در مجموع، 1200 مقاله شناسایی شد که پس از حذف موارد تکراری و غربالگری اولیه، 155 مقاله برای بررسی متن کامل باقی ماند و در نهایت 55 مطالعه در تحلیل وارد شد. دادهها بهصورت توصیفی و بر اساس نوع مداخله، مدل حیوانی و نتایج پیامدی ترکیب شدند.
یافتهها
مدلهای موشی:
برای درک مکانیسم ایجاد و گسترش بیماری پیوند علیه میزبان و همچنین ارزیابی اثربخشی روشهای درمانی مرتبط با این عارضه، از پیوند سلولهای انسانی در مدلهای حیوانی استفاده میشود. مدلهای موشهای دارای نقص ایمنی، به دلیل پیوند سلولها و بافتهای انسانی، «موشهای انسانی شده» نامیده میشوند. موشهای دیابتی غیر چاق با نقص ایمنی ترکیبی شدید از جمله اولین مدلهای موشی برای مطالعه بیماریهای انسانی مانند بیماری پیوند علیه میزبان بودند. با این حال، این موشها علیرغم نقص عملکرد سلولهای T و B، میزان پذیرش پیوند پایینی دارند که یکی از دلایل اصلی آن وجود سلولهای NK و پاسخ به آنتیژنهای بیگانه است. مدلهای دارای کمبود آنزیم RAG2 که فاقد عملکرد سلولهای T،B و NK هستند، پذیرش پیوند بالاتری نسبت به مدلهای قبلی داشتند. با این حال، در اوایل دهه ۲۰۰۰، توسعه مدلهای موشی دارای نقص ایمنی با جهش در زنجیره گاما اینترلوکین 2، منجر به پیشرفتهای عمدهای در مدلهای حیوانی شد. زنجیره گاما مشترک بخش مهمی از گیرندههای IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 است که برای اتصال قوی و ارتباط بین این سیتوکینها ضروری هستند (4-2).
موشهای دارای نقص ایمنی با جهش در IL-2R که امروزه به طور گسترده استفاده میشوند، شامل موشهای NOG ، BRG و NSG هستند. تفاوت مدلهای NOG و BRG با مدل NSG در این است که در هر دو مدل اول، زنجیره گاما گیرنده اینترلوکین 2 وجود دارد، اما مسیر سیگنالدهی آن مختل شده است، در حالی که در مدل NSG این زنجیره به طور کامل وجود ندارد. همه این موشها فاقد سلولهای لنفاوی (T, B, NK) هستند و سلولهای دندریتیک و ماکروفاژ آنها نیز فاقد عملکرد طبیعی هستند. بنابراین، استفاده از این مدلهای موشی برای درک و مطالعه مکانیسمهای القا، گسترش، کنترل و مهار بیماری پیوند علیه میزبان بسیار ارزشمند است (6-4).
استراتژیهای کاهش GvHD در مدلهای موشی:
علیرغم پیشرفتهای کلی در درمان و کاهش GvHD در سالهای اخیر، این بیماری هنوز یکی از مهمترین عوارض پیوند سلولهای بنیادی آلوژنیک است. از طرفی اثر پیوند بر سلول تومور/ لوسمی (Graft Versus Tumor/Leukemia, GVT/GVL) در موفقیت پیوند بسیار حائز اهمیت است. در GVT سلولهای ایمنی پیوند شده فقط بر سلولهای گیرنده بدخیم عمل میکنند و تومور را ریشهکن میکنند که نقش محافظتـی بیشتـری بــرای میزبـان ایفا میکند. امروزه از روشهای مختلفی برای کاهش GvHD بدون تأثیر بر GVT در مدلهای موشی که سلولهای انسانی دریافت کردهاند، استفاده میشود. داروهـای پیشگیری از GvHD، آنتیبادی ـ های مونوکلونال، مهارکنندههای سیتوکینهای التهابی و مهارکنندههای مسیر سیگنالینگ از جمله این روشها هستند (8، 7). در ادامه توضیح مختصری در مورد هر یک از این استراتژیها ارائه شده است.
داروهای پروفیلاکسی
- سیکلوسپورین A (Cyclosporine A):
سیکلوسپورین A (CyA) هنوز هم به عنوان رژیم اصلی سرکوبکننده سیستم ایمنی پس از پیوند مغز استخوان آلوژنیک استفاده میشود. سیکلوسپورین A برای اولین بار در سال 1982 در مدل حیوانی سگ آزمایش شد که اثرات مهاری بر aGvHD داشت. CyA با مهار آنزیم کلسینورین که منجر به مهار لنفوسیتهای T میشود، در کاهش GvHD و افزایش بقای بیماران مؤثر است (10، 9).
- آزتیدین (Azetidine):
آزتیدین (AZA) به عنوان یک عامل هیپومتیلاسیون در کاهش GvHD استفاده میشود. مدل موش NSG مبتلا به GvHD نشان داد که استفاده از AZA به طور کلی تکثیر لنفوسیتهای T انسانی را کاهش داده و تولید IFN-γ و TNF-α را کاهش میدهد. از سوی دیگر، کاهش گرانولهای گرانزیم و پرفورین نیز عملکرد سلولهای سیتوتوکسیک (cytotoxic T lymphocyte, CTL) را مختل میکند که در نهایت منجر به کاهش GvHD میشود (11). برخی مطالعهها نشان دادهاند که پیوند سلولهای مادر خونساز با جهش درDNMT3A (DNMT3A-Mutated HSC) به بیماران مبتلا به بدخیمیهای هماتولوژیک، علیرغم کاهش احتمال عود یا پیشرفت تومور، باعث افزایش خطر GvHD میشود (12).
- بورتزومیب (Bortezomib):
بورتزومیب رونویسی فاکتور هستهای NF-κB را مهار میکند و بنابراین تولید سیتوکینهای التهابی را کاهش میدهد (13).
سیتوکینها و کموکینها:
برای جلوگیری از طیف وسیعی از عوارض جانبی کورتیکواستروئیدها و ارائه یک گزینه درمانی برای بیماران GvHD که درمان کورتیکواستروئیدی آنها شکست خورده است، نقشهای متعدد سیتوکینها در پاتوفیزیولوژی aGvHD در یک مدل موش صحرایی بررسی شد. سیتوکینـ های التهابی ترشح شده توسط سلولهای T فعال، ماکروفاژها و سلولهای دندریتیک مانند TNF-α و IL-2 ، واسطههای التهابی کلیدی GvHD هستند و میتوانند اهداف درمانی مهمی در کاهش GvHD باشند. IL-6، IL-1، IL-11 و TNF-α در مدلهای موشی به عنوان اهدافی برای کاهش aGvHD مورد مطالعه قرار گرفته اند (14).
- اینترلوکینهای 11 و 35 (Interleukin 35 ، Interleukin 11):
IL-11 با قطبی کردن سلولهای T باعث تغییر این سلولها به سلولهای T کمکی نوع 2 (Th2) میشود که تأثیر کمتری در القای GvHD دارد. این سیتوکین همچنین با کاهش تولید IL-12 مرتبط بوده و در نتیجه مرگ و میر ناشی از GvHD را کاهش میدهد (15). مطالعهها در مدلهای موشی نشان داده است که این سیتوکین علیرغم این که به خوبی توسط موشها تحمل میشود، باعث عوارض جانبی شدید در انسان میگردد. مهمترین عارضه جانبی این سیتوکین، ادم ریوی است (16). از سوی دیگر، بیان IL-35 همچنین سلولهای T تنظیمی (Treg) را تقویت کرده و تولید سیتوکین از Th1 را سرکوب میکند که متعاقباً شدت GvHD را کاهش میدهد (17).
- آنالوگهای G-CSF (G-CSF Analogs) :
استفاده از آنالوگهای فاکتور رشد کلونی گرانولوسیت (G-CSF) تعداد و فعالیت سلولهای NK، تکثیر سلولهای Treg و فعال شدن سلولهای دندریتیک میزبان را افزایش میدهد. همچنین سلولهای CD4 مرتبط با GCD را مهار میکند. از طرف دیگر اثرات GVT وابسته به CD8 را افزایش میدهد (18).
- مهارکنندههای سیتوکینهای التهابی (Inflammatory Cytokines Inhibitors):
IL-1 به عنوان یک سیتوکین پیشالتهابی نقش مؤثری در ایجاد بیماری التهابی روده و آسیب بافتی دارد. این سیتوکین با اختلال در عملکرد سلولهای سرکوبگر مشتق از میلوئید (MDSC) شدت GvHD را در مدل موشی افزایش میدهد (19). استفاده از آنتاگونیستهای سیتوکین مانند آناکینرا شدت علائم GvHD را در مدلهای موشی کاهش داده است (20). IL-6 نیز یک سیتوکین فاز حاد است که استفاده از مسدودکنندههای آن مانند توکولیزوماب نیز شدت GvHD را در مدلهای موشی کاهش میدهد. مهارکنندههای IL-6 در ترکیب با مهارکنندههای TNF-α در بیماریهای خود ایمنی انسان مانند آرتریت روماتوئید استفاده میشوند (21). مطالعهها روی مدلهای موشی نشان داده است که TNF-α مسیر microRNA-146a/TRAF6 را تعدیل میکند و مستقیماً منجر به آسیب بافتی به سلولهای اپیتلیال روده میشود. همچنین عملکرد Treg را مهار کرده و شدت GvHD را افزایش میدهد. بنابراین استفاده از آنتاگونیست ـ های آن (اتانرسپت و اینفلیکسیماب) شدت GvHD را کاهش میدهد. با این حال، برخی مطالعهها نشان دادهاند که استفاده از این آنتاگونیستها، اثر GVT را در برابر برخی از سلولهای توموری کاهش میدهد (23، 22).
- مهارکننده IL-22 (Interleukin 22 inhibitors):
IL-22 توسـط سلولهــای Th17 (T helper 17) تـــولید
میشود که هم نقش محافظتی و هم نقش پیشالتهابی دارد. این سیتوکین همچنین نقش مهمی در پاتوفیزیولوژی GvHD در مدل موشی که پیوند سلولهای بنیادی آلوژنیک دریافت کرده است، ایفا میکند (24).
- کموکینها (Chemokines):
علاوه بر سیتوکینهایی که عملکرد سلولهای T را در GvHD تقویت میکنند، کموکینها همچنین میتوانند این سلولها را به سمت اندامهای هدف GvHD در مدل موش هدایت کنند. استفاده از مهارکنندههای کموکین بحث برانگیز است زیرا پرتو درمانی نیز مهاجرت سلولی را مختل میکند. با این حال، استفاده از مهارکنندههای CCR5 در مطالعههای اخیر نشان داده است که هیچ اثر محافظتی در کاهش شدت GvHD ندارد (25). همچنین، استفاده از آگونیست گیرنده اسفنگوزین 1 فسفات به نام فینگولیمود (FTY720) در مدلهای موشی، مهاجرت سلولهای T از غدد لنفاوی را مهار میکند و در نتیجه GvHD را کاهش میدهد (26).
آنتیبادیهای مونوکلونال:
استفاده از آنتیبادیها علیه سلولهــای T، Treg و B ، هدف اصلی از استراتژیهای مرتبط با آنتیبادی مونوکلونال برای کاهش GvHD هستند. بنابراین، آنتیبادیهای مونوکلونال علیه مارکرهای سطحی این سلولها برای درمان GvHD مورد مطالعه قرار گرفتهاند (27).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD26 :
CD26 نقش مهمی در مسیرهای سیگنالینگ فعالسـازی
لنفوسیت T، تعامل بین APC و لنفوسیتهای T و همچنین نقش تحریک همزمان روی سلول T پس از فعالسازی ایفا میکند. انتظار میرود مسدود کردن مولکول CD26 با استفاده از یک آنتیبادی مونوکلونال انسانی، با کاهش عملکرد CD8 باعث کاهش شدت GvHD شود. یک مطالعه در سال ۲۰۱۳ نشان داد که لنفوسیتهای CD26+ در اندام هدف GvHD وجود دارند. برای این منظور، پس از القای GvHD در موشها، سلولهای تک هستهای خون محیطی انسان به موشهای NOG تزریق شدند. علائم GvHD با لنفوسیتهای انسانی CD26+در خون محیطی و اندامهای هدف GvHD در این موشها یافت شد. متعاقباً، تزریق آنتیبادی مونوکلونال علیه CD26 ، شدت GvHD را کاهش داد و باعث افزایش طول عمر موش شد (28).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD28 :
مولکولهـای B7 در APC، فعالسـازی سلولهای T را از طریق سیگنالهـای فعالکننــده CD28 و مهــارکنندههای CTLA4 تنظیم مــیکنند.
از آن جایی که فعال شدن لنفوسیتهای T دهنده باعث GvHD میشود، سلولهای T دهنده فاقد CD28 در مقایسه با سلولهای T دهنده طبیعی، شدت GvHD را کاهش میدهند. نتایج مطالعهها نشان دادهاند که آنتیبادی ضد CD28 در جلوگیری از GvHD بهتر از CTLA4-Ig عمل میکند. این آنتیبادی مونوکلونال، برهمکنش CD28 با B7 را مهار میکند. این آنتیبادی همچنین ممکن است سیگنال تحریک همزمان CD28 از TCR/Ag را تضعیف کند (29).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن OX40L :
مولکول OX40L که بر سطح لنفوسیتهای B ، DC و اندوتلیال فعال بیان میشود، لیگاند CD134 (OX40) بر سطح لنفوسیتهای T فعال است. مطالعهها نشان دادهاند که اتصال OX40L به OX40 باعث GvHD میشود و استفاده از آنتاگونیست OX40L، GvHD را کاهش میدهد. در مطالعهای توسط بلازار در مدلهای موشی، نشان داده شد که تزریق لنفوسیتهای CD4 یا CD8 بدون OX40L به موشهای bm12 یا bm1، باعث فعال شدن لنفوسیت های T بافت پیوندی شده و با کاهش GvHD بدون تغییر اثر GVL همراه است (30).
- مهارکنندههای لنفوسیت T- آنتیبادی علیه آنتـیژن CD4 :
گلیکوپروتئین CD4 بر سطح سلولهای T کمکی، سلولهای دندریتیک و ماکروفاژها، پیوند ایجاد شده بین سلول T و APC را تقویت کرده و هنگام مواجهه با آنتی ژن، لنفوسیت T را فعال می کند. از این رو به آن گیرنده کمکی میگویند. در مطالعهای در سال ۲۰۱۴ توسط فریک و همکاران، GvHD در مدل موش BALB/Cwt که سلولهای طحال و مغز استخوان را از مدل موش TTG-C57Bl/6 دریافت کرده بوده، القا شد. اما موشهایی که آنتیبادیهای CD4 دریافت کردند، کاهش شدت GvHD و افزایش بقا را نشان دادند (31).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD83 :
مولکول CD83 در بلوغ تیموس، عملکرد محیطی و طول عمر لنفوسیتهای CD4 و همچنین بلوغ و عملکرد لنفوسیتهای B و سلولهای دندریتیک نقش دارد. از این رو، مولکول CD83 را میتوان یکی از استراتژیهای مهم برای جلوگیری از GvHD در نظر گرفت. در مطالعهای در سال ۲۰۱۳، وانگ و همکاران نشان دادند که در مدل موش(Severe Combined Immunodeficiency) SCID که لنفوسیتهای T انسانی دریافت کرده بود، تزریق آنتیبادی ضد CD83 القای GvHD را مهار میکند (32).
- مهارکنندههای لنفوسیت T- آنتیبادی علیه آنتیژن CD45RC :
CD45RC ایزوفرم متفاوتی از مولکولهای CD45 است که در بلوغ تیموسیت، فعالسازی و عملکرد لنفوسیتهای T مؤثر است. این نشانگر به میزان زیادی در سطح لنفوسیتهای B و T بیان میشود، در حالی که در سطح لنفوسیتهای تنظیمی FOXP3+ بیان نمیشود. در مطالعهای بر روی مدل موشی NSG ، مشاهده شد که تزریق آنتیبادی ضد مولکول CD45RC، GvHD را مهار میکند (33).
- مهارکنندههای لنفوسیت B- آنتیبادی علیه CD74 :
CD74 در سلولهای خونساز و غیر خونساز و سلولهای APC مانند سلولهای B و DC بیان میشود. مطالعهای در سال 2013 توسط چن و همکاران نشان داد که تزریق آنتیبادی CD74 به مدل موشی GvHD، تکثیر سلولهای آلوژنیک، تولید اینترفرون (IFN) و نفوذ لنفوسیتهای T را مهار میکند که در نهایت GvHD را کاهش میدهد (34).
- مهارکنندههای لنفوسیت T تنظیمی (Treg)- آنتیبادی علیه آنتیژن CD137 :
سلولهای T تنظیمی با فنوتیپ CD25/CD4 FOXP3+ نقش مهمی در تحمل ایمنی دارند. مطالعههای قبلی نشان دادهاند که سیگنال مولکول CD137، تکثیر و بقای سلولهای Treg را در شرایط آزمایشگاهی افزایش میدهد. بنابراین، سلولهای Treg همراه با آنتیبادیهای مونوکلونال ضد CD137 میتوانند ظرفیت سرکوب سیستم ایمنی را افزایش دهند. در مطالعهای در سال 2012 توسط کیم و همکاران، استفاده از آنتیبادی ضد CD137 به طور قابل توجهی GvHD حاد را در مدل موشی F1 (C57BL / 6.9 DBA / 2) مهار کرد. همچنین تعداد و عملکرد سلولهای Treg را در داخل بدن افزایش داد (35).
علاوه بر آنتی بادی های ذکر شده، استفاده از آنتیبادیهای مونوکلونال ضد FasL، CD132 و CD20 (ریتوکسیماب) نیز برای کاهش GvHD در مدلهای موشی استفاده شده است (27، 3).
مهارکنندههای مسیر سیگنالینگ:
سیتوکاینهای التهابی مانند IL-2، TNF-α و IL-17 از طریق اتصال به گیرنده خود، بر برخی مسیرهای سیگنالینگ، مانند مسیرهای سیگنالینگ Notch و JAK/STAT اثر میگذارند. این مسیرهای سیگنالینگ در بیان ژن، تعامل سلولی، تکثیر سلولی، فعالسازی سلولی و فرآیند پاتولوژیک GvHD نقش دارند. مهارکنندههای مختلفی که مولکولهای کلیدی در مسیرهای سیگنالینگ GvHD را هدف قرار میدهند، برای درمان GvHD مورد مطالعه قرار گرفتهاند (36، 3).
- مهارکنندههای مسیر JAK :
مولکولهای JAK نقش مهمی در پاسخهای التهابی در فرآیند القای GvHD دارند. 4 عضو این خانواده، تنظیم کنندههای اصلی عملکرد سلولهای ایمنی مانند DC ، T، B و نوتروفیلها هستند. از بین این چهار تنظیمکننده، JAK1 و JAK2 برای عملکرد و القای پاسخهای ایمنی سلول T ضروری هستند. مطالعهای در مدلهای موشی C57BL/6 (B6)، H-2Kb) و BALB/c (H-2Kd نشان داد که Ruxolitinib ، به عنوان مهارکننده JAK1 و JAK2 ، GvHD را کاهش میدهد. مکانیسم Ruxolitinib ، افزایش سلولهای Treg FOXP3+ و اختلال در تمایز لنفوسیتهای CD4 است که CD17 و IFNy تولید میکنند (38، 37).
- مهارکنندههای مسیر Notch :
سیگنالدهی Notch یک مسیر ارتباطی سلول ـ سلول است که نقش مهمی در توسعه و ایمنی سلولهای T ایفا میکند. مولکول DNMAML1 به عنوان یک مسدود کننده گیرنده فاکتور رونویسی Notch عمل میکند. DNMAML1 سلولهای Treg را افزایش میدهد، تولید سیتوکینهای التهابی را کاهش میدهد و فعالیت مسیرهای سیگنالدهی Ras/MAPK و NF-κB را کم میکند. DNMAML1 به طور قابل توجهی شدت GvHD را در مدلهای موشی کاهش میدهد (39).
- مهارکنندههای مسیر NF-κB :
سیگنالدهی NF-κB نقش بسیار مهمی در ایمنیزایی و تومورزایی دارد. c-Rel عضوی از خانواده NF-κB است. مطالعهای در سال 2014 توسط شونو در مدلهای موشی نشان داد که استفاده از مهارکنندههای c-Rel، GvHD را در عین حفظ اثر GVL کاهش میدهد. مکانیسم اصلی IT-901 (مهارکننده c-Rel)، کاهش واکنشپذیری و اختلال در بازخورد منفی تولید IL-2 است که منجر به تکثیر سلولهای Treg میشود. امروزه، IT-901 برای کاهش GvHD و درمان تومورهای لنفاوی استفاده میشود (40).
- مهارکنندههای مسیر STAT3 :
STAT3 یک فاکتور رونویسی سلولTh17 است که نقش مهمی در القای GvHD در مدلهای موشی ایفا میکند. PIAS3 به عنوان یک مهارکننده STAT3 به طور چشمگیری شدت و علائم بالینی aGvHD را در بافتهای هدف کبد، ریه، روده و پوست کاهش میدهد. مطالعهای در سال 2014 توسط سونگ هی لی در مدلهای موشی نشان داد که استفاده از مهارکننده PIAS3 (STAT3) شدت GvHD را کاهش داده و تولید سلولهای Th1 و Th17 را کاهش میدهد(41).
- مهارکننده فاکتـور هستهای فعالکننده لنفوسیت (NFAT) :
مهار هر دو فاکتور هستهای فعالکننده لنفوسیتهای T، NFAT-1 و NFAT-2 با مختل کردن تکثیر، مهاجرت و عملکرد لنفوسیتهای T، القای GvHD را کاهش میدهد (42).
علاوه بر مسیرهای سیگنالینگ ذکر شده، هدف قرار دادن سایر مسیرهای سیگنالینگ مانند PK-c PK-a ، MEK ، NFAT ، IRE-1a/XBP-1 ، Ikaros و غیره میتواند راهکارهایی برای کاهش GvHD باشد (36، 3).
اگر چه در مطالعههای مختلف، مسیرهای سیگنالینـــگ
درگیر در GvHD به صورت مستقل بررسی می شوند، اما در اصل این مسیرها به صورت یک شبکه هماهنگ عمل میکنند. پس از انجام پیوند سلولهای بنیادی، سلولهای APC میزبان فعال شده و مرحله آغازین پاسخ ایمنی را شکل میدهد. در این مرحله سیگنالهای TCR همراه کو محرکها مانند CD28 ، CD26 و OX40L منجر به فعالسازی و تکثیر کلونال سلولهای T و شروع طوفانی التهاب می شود (30-28). در ادامه مسیرهای CCR5 و S1P که FTY720 آن را هدف قرار میدهد، نقش تعیین کنندهای در هدایت سلولهای T بافت پیوندی از اندامهای لنفاوی به بافتهای هدف GvHD دارند. این مسیرها به صورت هماهنگ با مولکولهای چسبندگی، الگوی توزیع سلولهای T و در نتیجه شدت و محل درگیری ارگانها را مشخص میکنند (26، 25). در سطح سوم، مسیرهای تنظیمی مانند Treg، مولکولهای مهاری و تنظیمات متابولیک قرار دارند که تعادل بین سیگنالهای التهابی و ضد التهابی را کنترل میکنند (51). رویکردهای درمانی مبتنی بر سلول مانند سلولهای Treg و CD56+CD3+ و سلولهای مهندسی شده با تأثیر همزمان بر چند نقطه از این شبکه میتوانند توزیع سیتوکینها، میزان کو محرکها و نسبت سلولهای مؤثر و تنظیمی را تغییر داده و در نتیجه بر هر دو جنبه GvHD و GVL اثر بگذارند.
روشهای سلولی
با توجه به طول عمر کوتاه و ناکارآمدی داروهای کاهش دهنده GvHD، معمولاً از درمانهای سلولی برای کاهش GvHD استفاده میشود. سلولهایی مانند Treg، سلولهای دندریتیک مقاوم (TDCs)، سلولهای استرومایی مزانشیمی (MSCs) و غیره برای کاهش GvHD با حذف سلولهای T بافت پیوندی استفاده میشوند (36، 3).
- سلولهای ایمنی تنظیمی- سلول T تنظیمی (Treg):
سلولهای T تنظیمی علاوه بر تولید سیتوکینهای مهاری مانند IL-10 ، فعالیت طیف وسیعی از انواع سلولها، از جمله B، T، APC و NK را نیز سرکوب میکنند. نقش Treg در مهار GvHD در انسان و مدلهای موشی در مطالعههای قبلی نشان داده شده است. سلولهای Treg به شدت از تقسیم، تکثیر و تمایز سلولهای T دهنده جلوگیری میکنند که میتواند به عنوان یک درمان بالقوه برای هر دو نوع حاد و مزمن GvHD استفاده شود. تزریق Tregهای دهنده میتواند با موفقیت از aGVHD در موشها جلوگیری کند. در یک مطالعه، نشان داده شده است که انتقال سلولهای Treg FOXP3+ با کاهش GvHD در مدل موشی مرتبط است (43). در مطالعه دیگری، تزریق سلولهای Treg همچنین میتواند شروع GvHD را بدون ایجاد سمیت یا مرگ در مدلهای موشی NSG به تأخیر بیندازد (44).
- سلول TDC (Tolerance Dendritic Cell, TDC):
سلول TDC نقش مهمی در تحمل مرکزی و محیطی سیستم ایمنی ایفا میکند. این سلولها سطوح بالایی از سیتوکینهای مهاری ترشح میکنند. TDCها با تکثیر سلولهای Treg FOXP3+ و سرکوب تکثیر سلولهای CD4 مؤثر، القای GvHD را کاهش داده و بقا را افزایش میدهد (45).
- سلولهای استرومایی مزانشیمی (Mesenchymal Stromal Cell, MSC):
MSCs به عنوان یک جمعیت سلولی ناهمگن که در بسیاری از بافتها وجود دارند، ویژگیهای متفاوتی در سیستم ایمنی دارند. این سلولها در کنار هم، سلولهای ایمنی ذاتی مانند NK ، DC و ماکروفاژها و همچنین سلولهای ایمنی اکتسابی مانند T و B را کنترل و مهار میکنند. سلول های MSC میتوانند با تولید ROS ، NO ، آرژیناز 1، TGF-β و IL-10، فعالسازی، تکثیر و عملکرد سلولهای T را مهار کنند. مطالعههای In-vivo نشان دادهاند که این سلولها با القای آپوپتوز وابسته به پرفورین در سلولهای CD4، سرکوب قطبی شدن سلولهای Th1 و Th17 و افزایش شیفت به Th2، القای GvHD را کاهش میدهند (46).
- سلولهای سرکوبگر مشتق از میلوئید (Myeloid-derived Suppressor Cells, MDSCs):
MDSCs گروهی ناهمگن از سلولهای سرکوبگر میلوئیدی سیستم ایمنی هستند که از طریق تولید IL-10 ، تولید سلولهای Treg را القا کرده و ماکروفاژها را به فنوتیپ نوع 2 تغییر میدهند. همچنین میتوانند با تولید ترکیباتی از جمله NO ، ROS ، آرژیناز 1، TGF-β و IL-10 سلولهای T را مهار کنند. مطالعهای در یک مدل موش نیز نشان داد که سلولهای MDSC تولیدکننده IL-13 با مهار تکثیر و فعالسازی سلولهای T دهنده، GvHD را کاهش میدهند (47).
اخیراً، انتقال سلولهای انتخابی مانند سلولهای T DLI، CTL، NK و CAR از دیگر استراتژیهای سلولی برای کاهش GVHD در عین حفظ اثر GVL است (36).
- سلول کشنده طبیعی (Natural Killer cells, NK):
مطالعهای توسط اولسون و همکاران نشان داد که تزریق سلولهای NK در مدل موش GvHD با کاهش القای GvHD همراه است. (48) مطالعه دیگری توسط سونگ و همکاران در سال 2018 در مدل موش GvHD نشان داد که تزریق سلولهای NK فعال شده توسط IL-12 و IL-18 سلولهای T موثر را حذف کرده و در نتیجه GvHD را کاهش میدهد (49).
- سلولهای CAR T :
مطالعهای در سال 2017 توسط گوش و همکاران در یک مدل موش نشان داد که تزریق CAR آلوژنیک CD19 با کمک محرکهای CD28 ، فعالیت ضد لنفوم را با حداقل GvHD افزایش میدهد.
جهت انجام این مطالعه مروری، جست و جوی منابع در پایگاههای PubMed ،Scopus و Web of Science با استفاده از کلید واژههای مرتبط با GvHD، مدلهای حیوانی موش، مهارکنندههای سیتوکینـی، آنتــیبادیهای مــونوکلونال و مسیرهای سیگنالینگ، در بازه زمانی2000 تا 2025 انجام گرفت. معیارهای ورود شامل مطالعههای انگلیسی زبان دارای متن کامل بود که به بررسی مکانیسمهای ایجاد یا راهکارهای کاهش GvHD در مدلهای حیوانی، به ویژه موش، پرداخته بودند. مقالاتی که صرفاً بر مدل انسانی یا کشت سلولی متمرکز بودند، مقالات مروری و متاآنالیزها، و گزارشهای ناقص از مطالعه حذف شدند. در مجموع، 1200 مقاله شناسایی شد که پس از حذف موارد تکراری و غربالگری اولیه، 155 مقاله برای بررسی متن کامل باقی ماند و در نهایت 55 مطالعه در تحلیل وارد شد. دادهها بهصورت توصیفی و بر اساس نوع مداخله، مدل حیوانی و نتایج پیامدی ترکیب شدند.
یافتهها
مدلهای موشی:
برای درک مکانیسم ایجاد و گسترش بیماری پیوند علیه میزبان و همچنین ارزیابی اثربخشی روشهای درمانی مرتبط با این عارضه، از پیوند سلولهای انسانی در مدلهای حیوانی استفاده میشود. مدلهای موشهای دارای نقص ایمنی، به دلیل پیوند سلولها و بافتهای انسانی، «موشهای انسانی شده» نامیده میشوند. موشهای دیابتی غیر چاق با نقص ایمنی ترکیبی شدید از جمله اولین مدلهای موشی برای مطالعه بیماریهای انسانی مانند بیماری پیوند علیه میزبان بودند. با این حال، این موشها علیرغم نقص عملکرد سلولهای T و B، میزان پذیرش پیوند پایینی دارند که یکی از دلایل اصلی آن وجود سلولهای NK و پاسخ به آنتیژنهای بیگانه است. مدلهای دارای کمبود آنزیم RAG2 که فاقد عملکرد سلولهای T،B و NK هستند، پذیرش پیوند بالاتری نسبت به مدلهای قبلی داشتند. با این حال، در اوایل دهه ۲۰۰۰، توسعه مدلهای موشی دارای نقص ایمنی با جهش در زنجیره گاما اینترلوکین 2، منجر به پیشرفتهای عمدهای در مدلهای حیوانی شد. زنجیره گاما مشترک بخش مهمی از گیرندههای IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 است که برای اتصال قوی و ارتباط بین این سیتوکینها ضروری هستند (4-2).
موشهای دارای نقص ایمنی با جهش در IL-2R که امروزه به طور گسترده استفاده میشوند، شامل موشهای NOG ، BRG و NSG هستند. تفاوت مدلهای NOG و BRG با مدل NSG در این است که در هر دو مدل اول، زنجیره گاما گیرنده اینترلوکین 2 وجود دارد، اما مسیر سیگنالدهی آن مختل شده است، در حالی که در مدل NSG این زنجیره به طور کامل وجود ندارد. همه این موشها فاقد سلولهای لنفاوی (T, B, NK) هستند و سلولهای دندریتیک و ماکروفاژ آنها نیز فاقد عملکرد طبیعی هستند. بنابراین، استفاده از این مدلهای موشی برای درک و مطالعه مکانیسمهای القا، گسترش، کنترل و مهار بیماری پیوند علیه میزبان بسیار ارزشمند است (6-4).
استراتژیهای کاهش GvHD در مدلهای موشی:
علیرغم پیشرفتهای کلی در درمان و کاهش GvHD در سالهای اخیر، این بیماری هنوز یکی از مهمترین عوارض پیوند سلولهای بنیادی آلوژنیک است. از طرفی اثر پیوند بر سلول تومور/ لوسمی (Graft Versus Tumor/Leukemia, GVT/GVL) در موفقیت پیوند بسیار حائز اهمیت است. در GVT سلولهای ایمنی پیوند شده فقط بر سلولهای گیرنده بدخیم عمل میکنند و تومور را ریشهکن میکنند که نقش محافظتـی بیشتـری بــرای میزبـان ایفا میکند. امروزه از روشهای مختلفی برای کاهش GvHD بدون تأثیر بر GVT در مدلهای موشی که سلولهای انسانی دریافت کردهاند، استفاده میشود. داروهـای پیشگیری از GvHD، آنتیبادی ـ های مونوکلونال، مهارکنندههای سیتوکینهای التهابی و مهارکنندههای مسیر سیگنالینگ از جمله این روشها هستند (8، 7). در ادامه توضیح مختصری در مورد هر یک از این استراتژیها ارائه شده است.
داروهای پروفیلاکسی
- سیکلوسپورین A (Cyclosporine A):
سیکلوسپورین A (CyA) هنوز هم به عنوان رژیم اصلی سرکوبکننده سیستم ایمنی پس از پیوند مغز استخوان آلوژنیک استفاده میشود. سیکلوسپورین A برای اولین بار در سال 1982 در مدل حیوانی سگ آزمایش شد که اثرات مهاری بر aGvHD داشت. CyA با مهار آنزیم کلسینورین که منجر به مهار لنفوسیتهای T میشود، در کاهش GvHD و افزایش بقای بیماران مؤثر است (10، 9).
- آزتیدین (Azetidine):
آزتیدین (AZA) به عنوان یک عامل هیپومتیلاسیون در کاهش GvHD استفاده میشود. مدل موش NSG مبتلا به GvHD نشان داد که استفاده از AZA به طور کلی تکثیر لنفوسیتهای T انسانی را کاهش داده و تولید IFN-γ و TNF-α را کاهش میدهد. از سوی دیگر، کاهش گرانولهای گرانزیم و پرفورین نیز عملکرد سلولهای سیتوتوکسیک (cytotoxic T lymphocyte, CTL) را مختل میکند که در نهایت منجر به کاهش GvHD میشود (11). برخی مطالعهها نشان دادهاند که پیوند سلولهای مادر خونساز با جهش درDNMT3A (DNMT3A-Mutated HSC) به بیماران مبتلا به بدخیمیهای هماتولوژیک، علیرغم کاهش احتمال عود یا پیشرفت تومور، باعث افزایش خطر GvHD میشود (12).
- بورتزومیب (Bortezomib):
بورتزومیب رونویسی فاکتور هستهای NF-κB را مهار میکند و بنابراین تولید سیتوکینهای التهابی را کاهش میدهد (13).
سیتوکینها و کموکینها:
برای جلوگیری از طیف وسیعی از عوارض جانبی کورتیکواستروئیدها و ارائه یک گزینه درمانی برای بیماران GvHD که درمان کورتیکواستروئیدی آنها شکست خورده است، نقشهای متعدد سیتوکینها در پاتوفیزیولوژی aGvHD در یک مدل موش صحرایی بررسی شد. سیتوکینـ های التهابی ترشح شده توسط سلولهای T فعال، ماکروفاژها و سلولهای دندریتیک مانند TNF-α و IL-2 ، واسطههای التهابی کلیدی GvHD هستند و میتوانند اهداف درمانی مهمی در کاهش GvHD باشند. IL-6، IL-1، IL-11 و TNF-α در مدلهای موشی به عنوان اهدافی برای کاهش aGvHD مورد مطالعه قرار گرفته اند (14).
- اینترلوکینهای 11 و 35 (Interleukin 35 ، Interleukin 11):
IL-11 با قطبی کردن سلولهای T باعث تغییر این سلولها به سلولهای T کمکی نوع 2 (Th2) میشود که تأثیر کمتری در القای GvHD دارد. این سیتوکین همچنین با کاهش تولید IL-12 مرتبط بوده و در نتیجه مرگ و میر ناشی از GvHD را کاهش میدهد (15). مطالعهها در مدلهای موشی نشان داده است که این سیتوکین علیرغم این که به خوبی توسط موشها تحمل میشود، باعث عوارض جانبی شدید در انسان میگردد. مهمترین عارضه جانبی این سیتوکین، ادم ریوی است (16). از سوی دیگر، بیان IL-35 همچنین سلولهای T تنظیمی (Treg) را تقویت کرده و تولید سیتوکین از Th1 را سرکوب میکند که متعاقباً شدت GvHD را کاهش میدهد (17).
- آنالوگهای G-CSF (G-CSF Analogs) :
استفاده از آنالوگهای فاکتور رشد کلونی گرانولوسیت (G-CSF) تعداد و فعالیت سلولهای NK، تکثیر سلولهای Treg و فعال شدن سلولهای دندریتیک میزبان را افزایش میدهد. همچنین سلولهای CD4 مرتبط با GCD را مهار میکند. از طرف دیگر اثرات GVT وابسته به CD8 را افزایش میدهد (18).
- مهارکنندههای سیتوکینهای التهابی (Inflammatory Cytokines Inhibitors):
IL-1 به عنوان یک سیتوکین پیشالتهابی نقش مؤثری در ایجاد بیماری التهابی روده و آسیب بافتی دارد. این سیتوکین با اختلال در عملکرد سلولهای سرکوبگر مشتق از میلوئید (MDSC) شدت GvHD را در مدل موشی افزایش میدهد (19). استفاده از آنتاگونیستهای سیتوکین مانند آناکینرا شدت علائم GvHD را در مدلهای موشی کاهش داده است (20). IL-6 نیز یک سیتوکین فاز حاد است که استفاده از مسدودکنندههای آن مانند توکولیزوماب نیز شدت GvHD را در مدلهای موشی کاهش میدهد. مهارکنندههای IL-6 در ترکیب با مهارکنندههای TNF-α در بیماریهای خود ایمنی انسان مانند آرتریت روماتوئید استفاده میشوند (21). مطالعهها روی مدلهای موشی نشان داده است که TNF-α مسیر microRNA-146a/TRAF6 را تعدیل میکند و مستقیماً منجر به آسیب بافتی به سلولهای اپیتلیال روده میشود. همچنین عملکرد Treg را مهار کرده و شدت GvHD را افزایش میدهد. بنابراین استفاده از آنتاگونیست ـ های آن (اتانرسپت و اینفلیکسیماب) شدت GvHD را کاهش میدهد. با این حال، برخی مطالعهها نشان دادهاند که استفاده از این آنتاگونیستها، اثر GVT را در برابر برخی از سلولهای توموری کاهش میدهد (23، 22).
- مهارکننده IL-22 (Interleukin 22 inhibitors):
IL-22 توسـط سلولهــای Th17 (T helper 17) تـــولید
میشود که هم نقش محافظتی و هم نقش پیشالتهابی دارد. این سیتوکین همچنین نقش مهمی در پاتوفیزیولوژی GvHD در مدل موشی که پیوند سلولهای بنیادی آلوژنیک دریافت کرده است، ایفا میکند (24).
- کموکینها (Chemokines):
علاوه بر سیتوکینهایی که عملکرد سلولهای T را در GvHD تقویت میکنند، کموکینها همچنین میتوانند این سلولها را به سمت اندامهای هدف GvHD در مدل موش هدایت کنند. استفاده از مهارکنندههای کموکین بحث برانگیز است زیرا پرتو درمانی نیز مهاجرت سلولی را مختل میکند. با این حال، استفاده از مهارکنندههای CCR5 در مطالعههای اخیر نشان داده است که هیچ اثر محافظتی در کاهش شدت GvHD ندارد (25). همچنین، استفاده از آگونیست گیرنده اسفنگوزین 1 فسفات به نام فینگولیمود (FTY720) در مدلهای موشی، مهاجرت سلولهای T از غدد لنفاوی را مهار میکند و در نتیجه GvHD را کاهش میدهد (26).
آنتیبادیهای مونوکلونال:
استفاده از آنتیبادیها علیه سلولهــای T، Treg و B ، هدف اصلی از استراتژیهای مرتبط با آنتیبادی مونوکلونال برای کاهش GvHD هستند. بنابراین، آنتیبادیهای مونوکلونال علیه مارکرهای سطحی این سلولها برای درمان GvHD مورد مطالعه قرار گرفتهاند (27).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD26 :
CD26 نقش مهمی در مسیرهای سیگنالینگ فعالسـازی
لنفوسیت T، تعامل بین APC و لنفوسیتهای T و همچنین نقش تحریک همزمان روی سلول T پس از فعالسازی ایفا میکند. انتظار میرود مسدود کردن مولکول CD26 با استفاده از یک آنتیبادی مونوکلونال انسانی، با کاهش عملکرد CD8 باعث کاهش شدت GvHD شود. یک مطالعه در سال ۲۰۱۳ نشان داد که لنفوسیتهای CD26+ در اندام هدف GvHD وجود دارند. برای این منظور، پس از القای GvHD در موشها، سلولهای تک هستهای خون محیطی انسان به موشهای NOG تزریق شدند. علائم GvHD با لنفوسیتهای انسانی CD26+در خون محیطی و اندامهای هدف GvHD در این موشها یافت شد. متعاقباً، تزریق آنتیبادی مونوکلونال علیه CD26 ، شدت GvHD را کاهش داد و باعث افزایش طول عمر موش شد (28).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD28 :
مولکولهـای B7 در APC، فعالسـازی سلولهای T را از طریق سیگنالهـای فعالکننــده CD28 و مهــارکنندههای CTLA4 تنظیم مــیکنند.
از آن جایی که فعال شدن لنفوسیتهای T دهنده باعث GvHD میشود، سلولهای T دهنده فاقد CD28 در مقایسه با سلولهای T دهنده طبیعی، شدت GvHD را کاهش میدهند. نتایج مطالعهها نشان دادهاند که آنتیبادی ضد CD28 در جلوگیری از GvHD بهتر از CTLA4-Ig عمل میکند. این آنتیبادی مونوکلونال، برهمکنش CD28 با B7 را مهار میکند. این آنتیبادی همچنین ممکن است سیگنال تحریک همزمان CD28 از TCR/Ag را تضعیف کند (29).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن OX40L :
مولکول OX40L که بر سطح لنفوسیتهای B ، DC و اندوتلیال فعال بیان میشود، لیگاند CD134 (OX40) بر سطح لنفوسیتهای T فعال است. مطالعهها نشان دادهاند که اتصال OX40L به OX40 باعث GvHD میشود و استفاده از آنتاگونیست OX40L، GvHD را کاهش میدهد. در مطالعهای توسط بلازار در مدلهای موشی، نشان داده شد که تزریق لنفوسیتهای CD4 یا CD8 بدون OX40L به موشهای bm12 یا bm1، باعث فعال شدن لنفوسیت های T بافت پیوندی شده و با کاهش GvHD بدون تغییر اثر GVL همراه است (30).
- مهارکنندههای لنفوسیت T- آنتیبادی علیه آنتـیژن CD4 :
گلیکوپروتئین CD4 بر سطح سلولهای T کمکی، سلولهای دندریتیک و ماکروفاژها، پیوند ایجاد شده بین سلول T و APC را تقویت کرده و هنگام مواجهه با آنتی ژن، لنفوسیت T را فعال می کند. از این رو به آن گیرنده کمکی میگویند. در مطالعهای در سال ۲۰۱۴ توسط فریک و همکاران، GvHD در مدل موش BALB/Cwt که سلولهای طحال و مغز استخوان را از مدل موش TTG-C57Bl/6 دریافت کرده بوده، القا شد. اما موشهایی که آنتیبادیهای CD4 دریافت کردند، کاهش شدت GvHD و افزایش بقا را نشان دادند (31).
- مهارکنندههای لنفوسیت T-آنتیبادی علیه آنتیژن CD83 :
مولکول CD83 در بلوغ تیموس، عملکرد محیطی و طول عمر لنفوسیتهای CD4 و همچنین بلوغ و عملکرد لنفوسیتهای B و سلولهای دندریتیک نقش دارد. از این رو، مولکول CD83 را میتوان یکی از استراتژیهای مهم برای جلوگیری از GvHD در نظر گرفت. در مطالعهای در سال ۲۰۱۳، وانگ و همکاران نشان دادند که در مدل موش(Severe Combined Immunodeficiency) SCID که لنفوسیتهای T انسانی دریافت کرده بود، تزریق آنتیبادی ضد CD83 القای GvHD را مهار میکند (32).
- مهارکنندههای لنفوسیت T- آنتیبادی علیه آنتیژن CD45RC :
CD45RC ایزوفرم متفاوتی از مولکولهای CD45 است که در بلوغ تیموسیت، فعالسازی و عملکرد لنفوسیتهای T مؤثر است. این نشانگر به میزان زیادی در سطح لنفوسیتهای B و T بیان میشود، در حالی که در سطح لنفوسیتهای تنظیمی FOXP3+ بیان نمیشود. در مطالعهای بر روی مدل موشی NSG ، مشاهده شد که تزریق آنتیبادی ضد مولکول CD45RC، GvHD را مهار میکند (33).
- مهارکنندههای لنفوسیت B- آنتیبادی علیه CD74 :
CD74 در سلولهای خونساز و غیر خونساز و سلولهای APC مانند سلولهای B و DC بیان میشود. مطالعهای در سال 2013 توسط چن و همکاران نشان داد که تزریق آنتیبادی CD74 به مدل موشی GvHD، تکثیر سلولهای آلوژنیک، تولید اینترفرون (IFN) و نفوذ لنفوسیتهای T را مهار میکند که در نهایت GvHD را کاهش میدهد (34).
- مهارکنندههای لنفوسیت T تنظیمی (Treg)- آنتیبادی علیه آنتیژن CD137 :
سلولهای T تنظیمی با فنوتیپ CD25/CD4 FOXP3+ نقش مهمی در تحمل ایمنی دارند. مطالعههای قبلی نشان دادهاند که سیگنال مولکول CD137، تکثیر و بقای سلولهای Treg را در شرایط آزمایشگاهی افزایش میدهد. بنابراین، سلولهای Treg همراه با آنتیبادیهای مونوکلونال ضد CD137 میتوانند ظرفیت سرکوب سیستم ایمنی را افزایش دهند. در مطالعهای در سال 2012 توسط کیم و همکاران، استفاده از آنتیبادی ضد CD137 به طور قابل توجهی GvHD حاد را در مدل موشی F1 (C57BL / 6.9 DBA / 2) مهار کرد. همچنین تعداد و عملکرد سلولهای Treg را در داخل بدن افزایش داد (35).
علاوه بر آنتی بادی های ذکر شده، استفاده از آنتیبادیهای مونوکلونال ضد FasL، CD132 و CD20 (ریتوکسیماب) نیز برای کاهش GvHD در مدلهای موشی استفاده شده است (27، 3).
مهارکنندههای مسیر سیگنالینگ:
سیتوکاینهای التهابی مانند IL-2، TNF-α و IL-17 از طریق اتصال به گیرنده خود، بر برخی مسیرهای سیگنالینگ، مانند مسیرهای سیگنالینگ Notch و JAK/STAT اثر میگذارند. این مسیرهای سیگنالینگ در بیان ژن، تعامل سلولی، تکثیر سلولی، فعالسازی سلولی و فرآیند پاتولوژیک GvHD نقش دارند. مهارکنندههای مختلفی که مولکولهای کلیدی در مسیرهای سیگنالینگ GvHD را هدف قرار میدهند، برای درمان GvHD مورد مطالعه قرار گرفتهاند (36، 3).
- مهارکنندههای مسیر JAK :
مولکولهای JAK نقش مهمی در پاسخهای التهابی در فرآیند القای GvHD دارند. 4 عضو این خانواده، تنظیم کنندههای اصلی عملکرد سلولهای ایمنی مانند DC ، T، B و نوتروفیلها هستند. از بین این چهار تنظیمکننده، JAK1 و JAK2 برای عملکرد و القای پاسخهای ایمنی سلول T ضروری هستند. مطالعهای در مدلهای موشی C57BL/6 (B6)، H-2Kb) و BALB/c (H-2Kd نشان داد که Ruxolitinib ، به عنوان مهارکننده JAK1 و JAK2 ، GvHD را کاهش میدهد. مکانیسم Ruxolitinib ، افزایش سلولهای Treg FOXP3+ و اختلال در تمایز لنفوسیتهای CD4 است که CD17 و IFNy تولید میکنند (38، 37).
- مهارکنندههای مسیر Notch :
سیگنالدهی Notch یک مسیر ارتباطی سلول ـ سلول است که نقش مهمی در توسعه و ایمنی سلولهای T ایفا میکند. مولکول DNMAML1 به عنوان یک مسدود کننده گیرنده فاکتور رونویسی Notch عمل میکند. DNMAML1 سلولهای Treg را افزایش میدهد، تولید سیتوکینهای التهابی را کاهش میدهد و فعالیت مسیرهای سیگنالدهی Ras/MAPK و NF-κB را کم میکند. DNMAML1 به طور قابل توجهی شدت GvHD را در مدلهای موشی کاهش میدهد (39).
- مهارکنندههای مسیر NF-κB :
سیگنالدهی NF-κB نقش بسیار مهمی در ایمنیزایی و تومورزایی دارد. c-Rel عضوی از خانواده NF-κB است. مطالعهای در سال 2014 توسط شونو در مدلهای موشی نشان داد که استفاده از مهارکنندههای c-Rel، GvHD را در عین حفظ اثر GVL کاهش میدهد. مکانیسم اصلی IT-901 (مهارکننده c-Rel)، کاهش واکنشپذیری و اختلال در بازخورد منفی تولید IL-2 است که منجر به تکثیر سلولهای Treg میشود. امروزه، IT-901 برای کاهش GvHD و درمان تومورهای لنفاوی استفاده میشود (40).
- مهارکنندههای مسیر STAT3 :
STAT3 یک فاکتور رونویسی سلولTh17 است که نقش مهمی در القای GvHD در مدلهای موشی ایفا میکند. PIAS3 به عنوان یک مهارکننده STAT3 به طور چشمگیری شدت و علائم بالینی aGvHD را در بافتهای هدف کبد، ریه، روده و پوست کاهش میدهد. مطالعهای در سال 2014 توسط سونگ هی لی در مدلهای موشی نشان داد که استفاده از مهارکننده PIAS3 (STAT3) شدت GvHD را کاهش داده و تولید سلولهای Th1 و Th17 را کاهش میدهد(41).
- مهارکننده فاکتـور هستهای فعالکننده لنفوسیت (NFAT) :
مهار هر دو فاکتور هستهای فعالکننده لنفوسیتهای T، NFAT-1 و NFAT-2 با مختل کردن تکثیر، مهاجرت و عملکرد لنفوسیتهای T، القای GvHD را کاهش میدهد (42).
علاوه بر مسیرهای سیگنالینگ ذکر شده، هدف قرار دادن سایر مسیرهای سیگنالینگ مانند PK-c PK-a ، MEK ، NFAT ، IRE-1a/XBP-1 ، Ikaros و غیره میتواند راهکارهایی برای کاهش GvHD باشد (36، 3).
اگر چه در مطالعههای مختلف، مسیرهای سیگنالینـــگ
درگیر در GvHD به صورت مستقل بررسی می شوند، اما در اصل این مسیرها به صورت یک شبکه هماهنگ عمل میکنند. پس از انجام پیوند سلولهای بنیادی، سلولهای APC میزبان فعال شده و مرحله آغازین پاسخ ایمنی را شکل میدهد. در این مرحله سیگنالهای TCR همراه کو محرکها مانند CD28 ، CD26 و OX40L منجر به فعالسازی و تکثیر کلونال سلولهای T و شروع طوفانی التهاب می شود (30-28). در ادامه مسیرهای CCR5 و S1P که FTY720 آن را هدف قرار میدهد، نقش تعیین کنندهای در هدایت سلولهای T بافت پیوندی از اندامهای لنفاوی به بافتهای هدف GvHD دارند. این مسیرها به صورت هماهنگ با مولکولهای چسبندگی، الگوی توزیع سلولهای T و در نتیجه شدت و محل درگیری ارگانها را مشخص میکنند (26، 25). در سطح سوم، مسیرهای تنظیمی مانند Treg، مولکولهای مهاری و تنظیمات متابولیک قرار دارند که تعادل بین سیگنالهای التهابی و ضد التهابی را کنترل میکنند (51). رویکردهای درمانی مبتنی بر سلول مانند سلولهای Treg و CD56+CD3+ و سلولهای مهندسی شده با تأثیر همزمان بر چند نقطه از این شبکه میتوانند توزیع سیتوکینها، میزان کو محرکها و نسبت سلولهای مؤثر و تنظیمی را تغییر داده و در نتیجه بر هر دو جنبه GvHD و GVL اثر بگذارند.
روشهای سلولی
با توجه به طول عمر کوتاه و ناکارآمدی داروهای کاهش دهنده GvHD، معمولاً از درمانهای سلولی برای کاهش GvHD استفاده میشود. سلولهایی مانند Treg، سلولهای دندریتیک مقاوم (TDCs)، سلولهای استرومایی مزانشیمی (MSCs) و غیره برای کاهش GvHD با حذف سلولهای T بافت پیوندی استفاده میشوند (36، 3).
- سلولهای ایمنی تنظیمی- سلول T تنظیمی (Treg):
سلولهای T تنظیمی علاوه بر تولید سیتوکینهای مهاری مانند IL-10 ، فعالیت طیف وسیعی از انواع سلولها، از جمله B، T، APC و NK را نیز سرکوب میکنند. نقش Treg در مهار GvHD در انسان و مدلهای موشی در مطالعههای قبلی نشان داده شده است. سلولهای Treg به شدت از تقسیم، تکثیر و تمایز سلولهای T دهنده جلوگیری میکنند که میتواند به عنوان یک درمان بالقوه برای هر دو نوع حاد و مزمن GvHD استفاده شود. تزریق Tregهای دهنده میتواند با موفقیت از aGVHD در موشها جلوگیری کند. در یک مطالعه، نشان داده شده است که انتقال سلولهای Treg FOXP3+ با کاهش GvHD در مدل موشی مرتبط است (43). در مطالعه دیگری، تزریق سلولهای Treg همچنین میتواند شروع GvHD را بدون ایجاد سمیت یا مرگ در مدلهای موشی NSG به تأخیر بیندازد (44).
- سلول TDC (Tolerance Dendritic Cell, TDC):
سلول TDC نقش مهمی در تحمل مرکزی و محیطی سیستم ایمنی ایفا میکند. این سلولها سطوح بالایی از سیتوکینهای مهاری ترشح میکنند. TDCها با تکثیر سلولهای Treg FOXP3+ و سرکوب تکثیر سلولهای CD4 مؤثر، القای GvHD را کاهش داده و بقا را افزایش میدهد (45).
- سلولهای استرومایی مزانشیمی (Mesenchymal Stromal Cell, MSC):
MSCs به عنوان یک جمعیت سلولی ناهمگن که در بسیاری از بافتها وجود دارند، ویژگیهای متفاوتی در سیستم ایمنی دارند. این سلولها در کنار هم، سلولهای ایمنی ذاتی مانند NK ، DC و ماکروفاژها و همچنین سلولهای ایمنی اکتسابی مانند T و B را کنترل و مهار میکنند. سلول های MSC میتوانند با تولید ROS ، NO ، آرژیناز 1، TGF-β و IL-10، فعالسازی، تکثیر و عملکرد سلولهای T را مهار کنند. مطالعههای In-vivo نشان دادهاند که این سلولها با القای آپوپتوز وابسته به پرفورین در سلولهای CD4، سرکوب قطبی شدن سلولهای Th1 و Th17 و افزایش شیفت به Th2، القای GvHD را کاهش میدهند (46).
- سلولهای سرکوبگر مشتق از میلوئید (Myeloid-derived Suppressor Cells, MDSCs):
MDSCs گروهی ناهمگن از سلولهای سرکوبگر میلوئیدی سیستم ایمنی هستند که از طریق تولید IL-10 ، تولید سلولهای Treg را القا کرده و ماکروفاژها را به فنوتیپ نوع 2 تغییر میدهند. همچنین میتوانند با تولید ترکیباتی از جمله NO ، ROS ، آرژیناز 1، TGF-β و IL-10 سلولهای T را مهار کنند. مطالعهای در یک مدل موش نیز نشان داد که سلولهای MDSC تولیدکننده IL-13 با مهار تکثیر و فعالسازی سلولهای T دهنده، GvHD را کاهش میدهند (47).
اخیراً، انتقال سلولهای انتخابی مانند سلولهای T DLI، CTL، NK و CAR از دیگر استراتژیهای سلولی برای کاهش GVHD در عین حفظ اثر GVL است (36).
- سلول کشنده طبیعی (Natural Killer cells, NK):
مطالعهای توسط اولسون و همکاران نشان داد که تزریق سلولهای NK در مدل موش GvHD با کاهش القای GvHD همراه است. (48) مطالعه دیگری توسط سونگ و همکاران در سال 2018 در مدل موش GvHD نشان داد که تزریق سلولهای NK فعال شده توسط IL-12 و IL-18 سلولهای T موثر را حذف کرده و در نتیجه GvHD را کاهش میدهد (49).
- سلولهای CAR T :
مطالعهای در سال 2017 توسط گوش و همکاران در یک مدل موش نشان داد که تزریق CAR آلوژنیک CD19 با کمک محرکهای CD28 ، فعالیت ضد لنفوم را با حداقل GvHD افزایش میدهد.
به طور کلی، دادههای مدلهای موشی نشان میدهد که میتوان از سلولهای CAR-T برای افزایش پاسخ ضد لوسمی استفاده کرد، اگرچه اثرات آن بر GvHD بحثبرانگیز است (50).
سلولهای CIK و NKT :
زیر گروههای CD56+CD3+ این سلولها، شبیه سلولهای Treg بوده و باعث کاهش GvHD میشود (51).
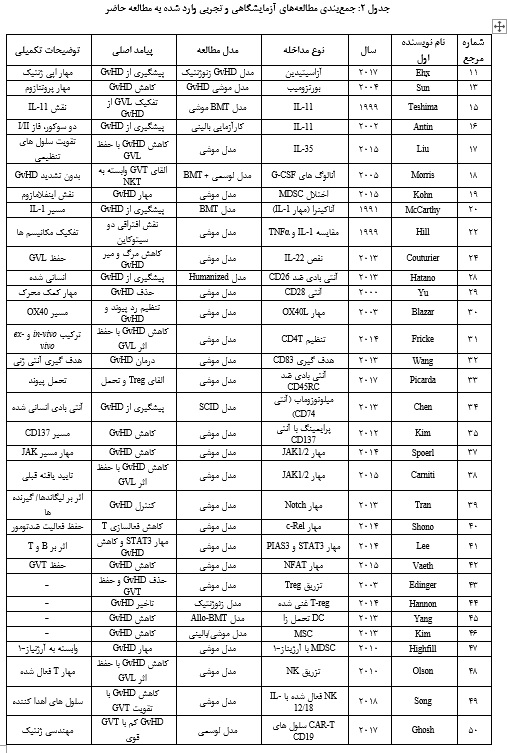
جمعبندی مطالعههای آزمایشگاهی و تجربی وارد شده به مطالعه حاضر در جدول ارائه شده است (جدول 2).
بحث
در این مطالعه، شواهد مربوط به مدل های حیوانی (موشی) GvHD و مداخلات ایمنی تنظیمی بررسی شد. نتایج نشان میدهد که پاتوژنز GvHD عمدتاً تحت تأثیر فعالسازی سلولهای T، مسیرهای التهابی و مهاجرت سلولهای ایمنی به اندامهای هدف قرار دارد. در این میان، مداخلاتی که مسیرهای فعال سازی یا ترافیک سلولهای ایمنی را هدف قرار میدهند، در کاهش شدت GvHD نتایج امیدوار کنندهای نشان دادهاند (شکل 2).
مدلهای موشی و مدلهای انسانی شده همچنان ابزارهای اصلی مطالعههای آزمایشگاهی GvHD محسوب میشوند. مدلهایی مانند NOG ، BRG و NSG در ترکیب با PBMC انسانی امکان بررسی دقیقتر مداخلات درمانی را فراهم میکنند؛ با این حال تفاوتهای بین گونهای، باعث محدودیت در پیش بینی کامل پاتولوژی GvHD در انسان میشود. از لحاظ مکانیسم اثر، مداخلات شناسایی شده در مطالعه حاضر را میتوان به سه گروه اصلی تقسیم کرد.
سلولهای CIK و NKT :
زیر گروههای CD56+CD3+ این سلولها، شبیه سلولهای Treg بوده و باعث کاهش GvHD میشود (51).
جمعبندی مطالعههای آزمایشگاهی و تجربی وارد شده به مطالعه حاضر در جدول ارائه شده است (جدول 2).
بحث
در این مطالعه، شواهد مربوط به مدل های حیوانی (موشی) GvHD و مداخلات ایمنی تنظیمی بررسی شد. نتایج نشان میدهد که پاتوژنز GvHD عمدتاً تحت تأثیر فعالسازی سلولهای T، مسیرهای التهابی و مهاجرت سلولهای ایمنی به اندامهای هدف قرار دارد. در این میان، مداخلاتی که مسیرهای فعال سازی یا ترافیک سلولهای ایمنی را هدف قرار میدهند، در کاهش شدت GvHD نتایج امیدوار کنندهای نشان دادهاند (شکل 2).
مدلهای موشی و مدلهای انسانی شده همچنان ابزارهای اصلی مطالعههای آزمایشگاهی GvHD محسوب میشوند. مدلهایی مانند NOG ، BRG و NSG در ترکیب با PBMC انسانی امکان بررسی دقیقتر مداخلات درمانی را فراهم میکنند؛ با این حال تفاوتهای بین گونهای، باعث محدودیت در پیش بینی کامل پاتولوژی GvHD در انسان میشود. از لحاظ مکانیسم اثر، مداخلات شناسایی شده در مطالعه حاضر را میتوان به سه گروه اصلی تقسیم کرد.
1) مهار فعالسازی سلولهای T مانند آنتیبادی ضد CD28 و CD26 که با کاهش هم تحریکی و پاسخ التهابی، GvHD را محدود میکنند. 2) مهار مهاجرت سلولی مانند CCR5 و FTY720 که بر مهاجرت سلولهای T اثر میگذارد؛ هر چند نتایج برای CCR5 مشابه و قانعکننده نبوده است. 3) مداخلات سلولی و ایمنی مانند CAR-T و CD56+CD3+ که بیشتر بر تعادل میان اثر GVL و کنترل GvHD تمرکز دارند. مقایسه مکانیسم اثر، مزایا و محدودیتها، تأثیر بر GvHD و GVL مداخلات ذکر شده در مطالعه در جدول 3 ارائه شده است. مطالعههای مختلف نشان دادهاند که هدف قرار دادن مسیرهای کلیدی فعالسازی سلولهای T، مهار سیتوکینهای التهابی و کنترل عملکرد سلولهای APC هم شدت GvHD را کاهش دادهاند و هم اثر GVL را حفظ کردهاند؛ حتی در برخی موارد GVL تقویت شده است (42، 38، 37، 24، 17، 13، 11).
در حوزه درمانهای سلولی نیز نتایج قابل توجهی گزارش شده است. در یک مدل موش، تزریق CAR آلوژنیک CD19 با کمک محرک CD28 موجب افزایش فعالیت GVL همراه با حداقل GvHD شده است، هر چند تأثیر دقیق آن بر GvHD همچنان مورد بحث است (50). همچنین گزارش شده است که سلولهایی با زیر جمعیت CD56+CD3+ میتوانند عملکردی مشابه Treg داشته باشند و از طریق مهار پاسخهای ایمنی، موجب کاهش GvHD شوند (51). در سالهای اخیر، رویکردهای نوین درمانی با تمرکز بر سلول درمانی، به ویژه سلولهای MSC و نیز هدفگیری مسیرهای سیگنالینگ مانند JAK/STAT با استفاده از مهارکنندههایی نظیر ruxolitinib ، نتایج امیدوارکنندهای در کاهش شدت GvHD و حفظ اثر GVT نشان دادهاند (54-52). علاوه بر مداخلاتی که توانستهاند در کاهش شدت GVHD مؤثر باشند، برخی مطالعهها نتایج منفی یا عوارض جانبی نشان دادهاند. به عنوان مثال، مهار مسیر CCR5 در برخی مدلها نتوانست اثر محافظتی قابلتوجهی در کاهش GvHD ایجاد کند که این موضوع میتواند به وجود مسیرهای جایگزین در مهاجرت و فعالسازی سلولهای T نسبت داده شود. این یافته نشان میدهد که مهار یک مسیر منفرد ممکن است برای کنترل کامل پاسخ التهابی کافی نباشد (25). از سوی دیگر، برخی روشهای درمانی مانند CAR‑T اگرچه اثر GVL قابلتوجهی دارند، اما در مورد تأثیر آنها بر GvHD نتایج یکنواختی گزارش نشده و در برخی موارد احتمال تشدید پاسخ ایمنی و افزایش التهاب سیستمیک وجود دارد (50). همچنین، مداخلاتی که مسیرهای کو محرکهای سلول T مانند CD28 را هدف قرار میدهند، در صورت مهار بیش از حد، ممکن است به کاهش شدید پاسخ ایمنی و ایجاد اختلال در تعادل ایمنی منجر شوند.
یکی از محدودیتهای تفسیر جامع نتایج مطالعههای آزمایشگاهی در مدلهای موشی، ناهمگونی مدلهای موشی پیوند است که میتواند به طور مستقیم بر شدت GvHD و پاسخ به مداخلات درمانی اثر بگذارد. تفاوتهای ژنتیکی میان دهنده و گیرنده، درجات متفاوتی از فعالسازی سلولهای T و در نتیجه شدتهای مختلف GvHD ایجاد میکنند. به علاوه، نوع نقص ایمنی گیرنده نیز عامل تعیین کنندهای است. مدلهای مبتنی بر اشعه با آسیب مخاطی، التهاب شدیدتری ایجاد میکنند؛ در حالی که مدلهای دارویی یا موشهای انسانی شده مانند NSG الگوی متفاوت و اغلب شدیدتری از GvHD نشان میدهند. این تفاوتها باعث تغییر اثربخشی مداخلات انجام شده میگردد. به طور کلی، این تفاوتها نشان میدهند که تفسیر نتایج درمانی باید با توجه به ویژگیهای ژنتیکی و ایمنی مدل استفاده شده انجام شود و تعمیم نتایج به سایر مدلها و انسان باید با احتیاط صورت گیرد.
نتیجهگیری
با توجه به بروز بالای عوارض و مرگومیر ناشی از GvHD، درک مکانیسمهای اصلی درگیر در این عارضه برای شناسایی راهکارهای درمانی مؤثر اهمیت دارد. در اغلب مدلهای آزمایشگاهی، ابتدا سلولهای PBMC انسانی به موشهای تحت تابش تزریق شده و سپس مداخلات مختلف برای کاهش GvHD ارزیابی شدهاند (38، 37). یکی از چالشهای اصلی، ایجاد تعادل میان مهار GvHD و حفظ اثر ضد توموری پیوند است؛ به طوری که برخی مداخلات مانند آزاتیدین و مهارکنندههای پروتئازوم توانستهاند هر دو هدف را همزمان حفظ کنند (38، 37). این تعادل تحت تأثیر نوع مداخله، زمان اعمال آن و نقش زیر جمعیتهای سلولهای T و سلولهای ایمنی ذاتی مانند NK و MDSC قرار دارد (38، 37). در برخی راهبردها مانند واکسیناسیون علیه WT1، افزایش اثر GVT همراه با کنترل GvHD مشاهده شده است (55، 49). همچنین، مدلهای انسانیشده مانند NSG، NOG و BRG ابزارهای مهمی برای مطالعه مکانیسمها و ارزیابی درمانهای نوظهور محسوب میشوند. شواهد نشان میدهد که مداخلات هدفمند علیه مسیرهای فعالسازی و مهاجرت سلولهای T بیشترین ظرفیت را برای کاهش شدت GvHD دارند، اگرچه اثر آنها بر حفظ GVL در مطالعههای مختلف متفاوت گزارش شده است. با وجود پیشرفتها، ناهمگونی در طراحی مطالعهها و محدودیتهای مدلهای حیوانی موش، تعمیمپذیری نتایج را کاهش میدهد. بهبود طراحی مدلها، افزایش استانداردسازی، بررسی ترکیب درمانها و توجه به مطالعههایی که نتایج منفی یا عوارض ناخواسته نشان دادهاند، میتواند به درک محدودیتهای هر روش و ارائه راهبردهای دقیقتر و مؤثرتر برای کنترل پایدار GvHD کمک کند (38، 37).
حمایت مالی
این پژوهش با حمایت معنوی دانشگاه علوم پزشکی بندرعباس انجام شده است.
نقش نویسندگان
دکتر فاطمه روشن ضمیر: نگارش نسخه نهایی و ویرایش نهایی مقاله
دکتر جواد مهاجر انصاری: ویرایش مقاله
دکتر مرضیه نوروزیان: نگارش مقاله
دکتر مجید ترماحی اردستانی: جمعآوری مطالب و نگارش نسخه اولیه مقاله
نوع مطالعه: مروري |
موضوع مقاله:
خون و انكولوژي
فهرست منابع
1. Teshima T, Reddy P, Zeiser R. Reprint of: acute graft-versus-host disease: novel biological insights. Biol Blood Marrow Transplant. 2016; 22(3): S3-8. [DOI:10.1016/j.bbmt.2016.01.004] [PMID]
2. Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis model mech. 2011; 4(3): 318-33. [DOI:10.1242/dmm.006668] [PMID] []
3. Zhang L, Yu J, Wei W. Advance in targeted immunotherapy for graft-versus-host disease. Front immunol. 2018; 9: 1087. [DOI:10.3389/fimmu.2018.01087] [PMID] []
4. Walsh NC, Kenney LL, Jangalwe S, Aryee K-E, Greiner DL, Brehm MA, et al. Humanized mouse models of clinical disease. Annu Rev Pathol. 2017; 12: 187-215. [DOI:10.1146/annurev-pathol-052016-100332] [PMID] []
5. Brehm M.A, Bortell R, Verma M, Shultz L. D, Greiner D. Humanized mice in translational immunology. Translational immunology: mechanisms and pharmacological approaches. 2016: 285-326. [DOI:10.1016/B978-0-12-801577-3.00012-5]
6. Hogenes MC. B cells and regulatory T cells in Graft versus Host Disease: a clinicopathological study in humanized mice: Utrecht University; 2019.
7. Hülsdünker J, Zeiser R. Insights into the pathogenesis of GvHD: what mice can teach us about man. Tissue Antigens. 2015; 85(1): 2-9. [DOI:10.1111/tan.12497] [PMID]
8. Zeiser R, Blazar BR. Preclinical models of acute and chronic graft-versus-host disease: how predictive are they for a successful clinical translation? Blood. 2016; 127(25): 3117-26. [DOI:10.1182/blood-2016-02-699082] [PMID] []
9. Deeg H.J, Storb R, Weiden P.L, Raff R.F, Sale G.E, Atkinson K, et al. Cyclosporin A and methotrexate in canine marrow transplantation: engraftment, graft-versus-host disease, and induction of intolerance. Transplantation. 1982; 34(1): 30-5. [DOI:10.1097/00007890-198207000-00006] [PMID]
10. Ardestani M.T, Kazemi A, Chahardouli B, Mohammadi S, Nikbakht M, Rostami Sh, et al. FLT3-ITD compared with DNMT3A R882 mutation is a more powerful independent inferior prognostic factor in adult acute myeloid leukemia patients after allogeneic hematopoietic stem cell transplantation: a retrospective cohort study. Turk J Haematol. 2018; 35(3): 158-67. [DOI:10.4274/tjh.2018.0017] [PMID] []
11. Ehx G, Fransolet G, De Leval L, D'Hondt S, Lucas S, Hannon M, et al. Azacytidine prevents experimental xenogeneic graft-versus-host disease without abrogating graft-versus-leukemia effects. Oncoimmunology. 2017; 6(5): e1314425. [DOI:10.1080/2162402X.2017.1314425] [PMID] []
12. Ardestani M.T, Norouzian M. A Review of Immune Landscape of Clonal Hematopoiesis: Progression and Prospects for the Future. Hormozgan Medical Journal. 2023 Oct 1; 27(4): 177-86. [DOI:10.34172/hmj.8122]
13. Sun K, Welniak L.A, Panoskaltsis-Mortari A, O'Shaughnessy M.J, Liu H, Barao I, et al. Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Sci USA. 2004; 101(21): 8120-5. [DOI:10.1073/pnas.0401563101] [PMID] []
14. Kumar S, Mohammadpour H, Cao X. Targeting cytokines in GVHD therapy. J Immunol Res Ther. 2017; 2(1): 90-9.
15. Teshima T, Hill G.R, Pan L, Brinson YS, Van Den Brink MR, Cooke KR, et al. IL-11 separates graft-versus-leukemia effects from graft-versus-host disease after bone marrow transplantation. J cli invest. 1999; 104(3): 317-25. [DOI:10.1172/JCI7111] [PMID] []
16. Antin JH, Lee SJ, Neuberg D, Alyea E, Soiffer RJ, Sonis S, et al. A phase I/II double-blind, placebo-controlled study of recombinant human interleukin-11 for mucositis and acute GVHD prevention in allogeneic stem cell transplantation. Bone marrow transplant. 2002; 29(5): 373-7. [DOI:10.1038/sj.bmt.1703394] [PMID]
17. Liu Y, Wu Y, Wang Y, Cai Y, Hu B, Bao G, et al. IL-35 mitigates murine acute graft-versus-host disease with retention of graft-versus-leukemia effects. Leukemia. 2015; 29(4): 939-46. [DOI:10.1038/leu.2014.310] [PMID] []
18. Morris ES, MacDonald KP, Rowe V, Banovic T, Kuns RD, Don AL, et al. NKT cell-dependent leukemia eradication following stem cell mobilization with potent G-CSF analogs. J Clin Invest. 2005; 115(11): 3093-103. [DOI:10.1172/JCI25249] [PMID] []
19. Koehn BH, Apostolova P, Haverkamp JM, Miller JS, McCullar V, Tolar J, et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood. 2015; 126(13): 1621-8. [DOI:10.1182/blood-2015-03-634691] [PMID] []
20. McCarthy Jr PL, Abhyankar S, Neben S, Newman G, Sieff C, Thompson RC, et al. Inhibition of interleukin-1 by an interleukin-1 receptor antagonist prevents graft-versus-host disease. Blood.1991; 78(8): 1915-8 [DOI:10.1182/blood.V78.8.1915.bloodjournal7881915] [PMID]
21. Kennedy GA, Varelias A, Vuckovic S, Le Texier L, Gartlan KH, Zhang P, et al. Addition of interleukin-6 inhibition with tocilizumab to standard graft-versus-host disease prophylaxis after allogeneic stem-cell transplantation: a phase 1/2 trial. Lancet Oncol. 2014; 15(13): 1451-9. [DOI:10.1016/S1470-2045(14)71017-4] [PMID]
22. Hill GR, Teshima T, Gerbitz A, Pan L, Cooke KR, Brinson YS, et al. Differential roles of IL-1 and TNF-α on graft-versus-host disease and graft versus leukemia. J Clin Invest. 1999; 104(4): 459-67. [DOI:10.1172/JCI6896] [PMID] []
23. Valencia X, Stephens G, Goldbach-Mansky R, Wilson M, Shevach EM, Lipsky PE. TNF downmodulates the function of human CD4+ CD25hi T-regulatory cells. Blood. 2006; 108(1): 253-61. [DOI:10.1182/blood-2005-11-4567] [PMID] []
24. Couturier M, Lamarthee B, Arbez J, Renauld J-C, Bossard C, Malard F, et al. IL-22 deficiency in donor T cells attenuates murine acute graft-versus-host disease mortality while sparing the graft-versus-leukemia effect. Leukemia. 2013; 27(7): 1527-37. [DOI:10.1038/leu.2013.39] [PMID]
25. Hammond WA, Heckman M, Finn L, Diehl NN, Shreders A, Flowers P, et al. No evidence of impact of maraviroc on outcome after allogeneic hematopoietic stem cell transplant with reduced intensity conditioning (RIC). Biol Blood Marrow Transplant. 2016; 22(3): S396-7. [DOI:10.1016/j.bbmt.2015.11.921]
26. Potì F, Gualtieri F, Sacchi S, Weißen-Plenz G, Varga G, Brodde M, et al. KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R−/− mice. Arterioscler Thromb Vasc Biol. 2013; 33(7): 1505-12. [DOI:10.1161/ATVBAHA.113.301347] [PMID]
27. Oelkrug C, Sack U, Boldt A, Nascimento IC, Ulrich H, Fricke S. Antibody‐and aptamer‐strategies for GvHD prevention. J Cell Mol Med. 2015; 19(1): 11-20. [DOI:10.1111/jcmm.12416] [PMID] []
28. Hatano R, Ohnuma K, Yamamoto J, Dang NH, Yamada T, Morimoto C. Prevention of acute graft‐versus‐host disease by humanized anti‐CD 26 monoclonal antibody. Br J Haematol. 2013; 162(2): 263-77. [DOI:10.1111/bjh.12378] [PMID]
29. Yu XZ, Bidwell SJ, Martin PJ, Anasetti C. CD28-specific antibody prevents graft-versus-host disease in mice. J Immunol. 2000 ; 164(9): 4564-8. [DOI:10.4049/jimmunol.164.9.4564] [PMID]
30. Blazar BR, Sharpe AH, Chen AI, Panoskaltsis-Mortari A, Lees C, Akiba H, et al. Ligation of OX40 (CD134) regulates graft-versus-host disease (GVHD) and graft rejection in allogeneic bone marrow transplant recipients. Blood. 2003; 101(9): 3741-8. [DOI:10.1182/blood-2002-10-3048] [PMID]
31. Fricke S, Hilger N, Fricke C, Schönfelder U, Behre G, Ruschpler P, et al. Prevention of graft-versus-host-disease with preserved graft-versus-leukemia-effect by ex vivo and in vivo modulation of CD4+ T-cells. Cell Mol Life Sci . 2014; 71(11): 2135-48. [DOI:10.1007/s00018-013-1476-0] [PMID] []
32. Wang X, Wei MQ, Liu X. Targeting CD83 for the treatment of graft-versus-host disease. Experimental and Therapeutic Medicine. 2013;5(6):1545-50. [DOI:10.3892/etm.2013.1033] [PMID] []
33. Picarda E, Bézie S, Boucault L, Autrusseau E, Kilens S, Meistermann D, et al. Transient antibody targeting of CD45RC induces transplant tolerance and potent antigen-specific regulatory T cells. JCI insight. 2017; 2(3): e90088. [DOI:10.1172/jci.insight.90088] [PMID] []
34. Chen X, Chang C-H, Stein R, Cardillo TM, Gold DV, Goldenberg DM. Prevention of acute graft-versus-host disease in a xenogeneic SCID mouse model by the humanized anti-CD74 antagonistic antibody milatuzumab. Biol Blood Marrow Transplant. 2013; 19(1): 28-39. [DOI:10.1016/j.bbmt.2012.09.015] [PMID]
35. Kim J, Kim W, Kim HJ, Park S, Kim H-A, Jung D, et al. Host CD25+ CD4+ Foxp3+ regulatory T cells primed by anti-CD137 mAbs inhibit graft-versus-host disease. Biol Blood Marrow Transplant. 2012; 18(1) :44-54. [DOI:10.1016/j.bbmt.2011.09.004] [PMID]
36. Chang Y-J, Zhao X-Y, Huang X-J. Strategies for enhancing and preserving anti-leukemia effects without aggravating graft-versus-host disease. Front Immunol. 2018 :21:3041. [DOI:10.3389/fimmu.2018.03041] [PMID] []
37. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014; 123(24) : 3832-42. [DOI:10.1182/blood-2013-12-543736] [PMID]
38. Carniti C, Gimondi S, Vendramin A, Recordati C, Confalonieri D, Bermema A, et al. Pharmacologic inhibition of JAK1/JAK2 signaling reduces experimental murine acute GVHD while preserving GVT effects. Clin Cancer Res. 2015; 21(16): 3740-9. [DOI:10.1158/1078-0432.CCR-14-2758] [PMID]
39. Tran IT, Sandy AR, Carulli AJ, Ebens C, Chung J, Shan GT, et al. Blockade of individual Notch ligands and receptors controls graft-versus-host disease. J Clin Invest. 2013; 123(4): 1590-604. [DOI:10.1172/JCI65477] [PMID] []
40. Shono Y, Tuckett AZ, Ouk S, Liou H-C, Altan-Bonnet G, Tsai JJ, et al. A small-molecule c-Rel inhibitor reduces alloactivation of T cells without compromising antitumor activity. Cancer Discov. 2014; 4(5): 578-91. [DOI:10.1158/2159-8290.CD-13-0585] [PMID] []
41. Lee S-H, Moon S-J, Park M-J, Kim E-K, Moon Y-M, Cho M-L. PIAS3 suppresses acute graft-versus-host disease by modulating effector T and B cell subsets through inhibition of STAT3 activation. Immunol Lett. 2014; 160(1): 79-88. [DOI:10.1016/j.imlet.2014.03.014] [PMID]
42. Vaeth M, Bäuerlein CA, Pusch T, Findeis J, Chopra M, Mottok A, et al. Selective NFAT targeting in T cells ameliorates GvHD while maintaining antitumor activity. Proc Natl Acad Sci USA. 2015; 112(4): 1125-30. [DOI:10.1073/pnas.1409290112] [PMID] []
43. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+ CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003; 9(9): 1144-50. [DOI:10.1038/nm915] [PMID]
44. Hannon M, Lechanteur C, Lucas S, Somja J, Seidel L, Belle L, et al. Infusion of clinical‐grade enriched regulatory T cells delays experimental xenogeneic graft‐versus‐host disease. Transfusion. 2014; 54(2): 353-63. [DOI:10.1111/trf.12279] [PMID]
45. Yang J, Li R, Ren Y, Yang Y, Xie R, Fan H. Third‐party tolerogenic dendritic cells reduce allo‐reactivity in vitro and ameliorate the severity of acute graft‐versus‐host disease in allo‐bone marrow transplantation. Scand J Immunol.2013; 78(6): 486-96. [DOI:10.1111/sji.12113] [PMID]
46. Kim N, Im K-I, Lim J-Y, Jeon E-J, Nam Y-S, Kim E-J, et al. Mesenchymal stem cells for the treatment and prevention of graft-versus-host disease: experiments and practice. Ann Hematol. 2013; 92(10): 1295-308. [DOI:10.1007/s00277-013-1796-z] [PMID]
47. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood. 2010; 116(25): 5738-47. [DOI:10.1182/blood-2010-06-287839] [PMID] []
48. Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010; 115(21): 4293-301. [DOI:10.1182/blood-2009-05-222190] [PMID] []
49. Song Y, Hu B, Liu Y, Jin Z, Zhang Y, Lin D, et al. IL‐12/IL‐18‐preactivated donor NK cells enhance GVL effects and mitigate GvHD after allogeneic hematopoietic stem cell transplantation. Eur J Immunol. 2018; 48(4): 670-82. [DOI:10.1002/eji.201747177] [PMID]
50. Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017; 23(2): 242-9. [DOI:10.1038/nm.4258] [PMID] []
51. Guo Y, Han W. Cytokine-induced killer (CIK) cells: from basic research to clinical translation. Chin J Cancer. 2015; 34(3): 99-107. [DOI:10.1186/s40880-015-0002-1] [PMID] []
52. Han X , Liao R, Li X, Zhang C, Huo SH,Qin L,et al. Mesenchymal stem cells in treating human diseases: molecular mechanisms and clinical studies. Signal Transduct Target Ther. 2025; 10(1): 262. [DOI:10.1038/s41392-025-02313-9] [PMID] []
53. Leonard JD, Peterson P. JAK inhibition immunotherapy for APS-1. N Engl J Med. 2024; 390(11): 1045 1047. [DOI:10.1056/NEJMe2403419] [PMID]
54. Jabbour E, Verstovsek S. Ruxolitinib. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK570600/
55. Rostami S, Kazemi A, Chahardouli B, Mohammadi S, Nikbakht M, Alizadeh N, et al. The prognostic impact of WT1 expression levels, mutations, and SNP rs16754 in AML patients: A retrospective cohort study. J Adv Med Biomed Res. 2021 Feb 10; 29(133): 109-17.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
