

جلد 19، شماره 4 - ( زمستان 1401 )
جلد 19 شماره 4 صفحات 312-301 |
برگشت به فهرست نسخه ها

![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Hadad deilami A, Salehi M, Shahabi M. Cloning and expression a-galactosidase in order to convert B to O blood group. bloodj 2022; 19 (4) :301-312
URL: http://bloodjournal.ir/article-1-1450-fa.html
URL: http://bloodjournal.ir/article-1-1450-fa.html
حداد دیلمی افسانه، صالحی میترا، شهابی مجید. کلونینگ آنزیم آلفا گالاکتوزیداز با هدف تبدیل آنتیژن B گروه خونی به آنتیژن O. فصلنامه پژوهشی خون. 1401; 19 (4) :301-312



استادیار دانشکده علم زیستی دانشگاه آزاد اسلامی واحد تهران شمال
متن کامل [PDF 597 kb]
(911 دریافت)
| چکیده (HTML) (1932 مشاهده)
مقدمه
سیستم گروه خونی ABO مهمترین سیستم گروه خونی در طب انتقال خون میباشد. پایه آنتیژنهای این سیستم ماده H میباشد که در سطح تمام گلبولهای قرمز وجود دارد. جایگاه ژنی ABO داری سه آلل A ، B و O میباشد(1).
آللهای A و B آنزیمهای ترانسفرازی را کد میکنند که باعث اتصال واحدهای کربوهیدراتی به ماده H میشوند. محصول پروتئینی آلل A آنزیم ان استیل گالاکتوزآمین ترانسفراز است که یک مولکول ان استیل گالاکتوزآمین را به ماده H متصل میکند و آنتیژن A را به وجود می آورد. آلل Bآنزیم گالاکتوزیل ترانسفراز را کد میکند که با افزودن یک مولکول گالاکتوز به ماده Hآن را تبدیل به آنتیژن B میکند. آلل O فاقد محصول خاصی بوده و افراد هموزیگوت برای این آلل، ماده H را به صورت دست نخورده در سطح گلبولهای قرمز خود بیان میکنند.
به دلیل وجود آنتیبادیهای ABO در همه افراد، بدون مواجهه قبلی با گلبولهای قرمز انسانی، گروهبندی صحیح اهداکننده و دریافتکننده خون از نظر ABO ، اساس ایمنی انتقال خون را تشکیل میدهد. تزریق خون ناسازگار از نظر ABO به بیمار میتواند منجر به همولیز داخل عروقی و عوارض جدی ناشی از واکنش انتقال خون حاد شود(3، 2).
در بین گروههای خونی، گروه O به عنوان دهنده همگانی برای بسیاری از بیماران به ویژه در موارد اورژانسی که امکان تعیین گروه ABO میسر نیست مورد استفاده قرار میگیرد(4). بررسی فراوانی گروههای ABO در جمعیت ایرانی نشان داده که گروه O و A بیشترین درصد جمعیت را تشکیل میدهند(به ترتیب 49/36 و 09/32 درصد). به دنبال آنها گروه B با فراوانی 68/23% و گروه AB با فراوانی 74/7% قرار دارند(5).
اندیشه تبدیل همه گروههای سیستم ABOبه گروه O از ابتدای دهه 1990میلادی مطرح شد. در بین گروههای خونی، آنتیژن B فقط در گالاکتوز انتهایی با آنتیژن O متفاوت است. بنابراین با استفاده از آنزیم گالاکتوزیداز (α-galactosidas)، میتوان گروه B را به O تبدیل کرد(8-6). از آن جا که ناخالصیهای همراه با آنزیم استخراج شده از منابع گیاهی میتوانست منجر به بروز عوارض و واکنشهای ناخواسته شود. از این رو تکنولوژی DNA نوترکیب مورد استفاده گرفت. استفاده از این تکنولوژی، امکان تولید آنزیم را در مقیاس وسیع و با آلودگی کمتر امکانپذیر ساخته است. به علاوه، مشخص شده که نوع نوترکیب در مقایسه با آلفاگالاکتوزیداز استخراج شده از منابع گیاهی(دانه سبز قهوه) بسیار کارآمدتر است(10، 9).
در صورتی که امکان تبدیل خون افراد گروه B به گروه O میسر شود، گام مهمی در جهت فراهم آوردن ذخیره خونی میباشد. یکی از مشکلات در این زمینه تولید انبوه آنزیم آلفاگالاکتوزیداز با کارآیی مناسب و در مقیاس وسیع است.
لذا مطالعه حاضر، با هدف تولید آنزیم بهینهسازی شده آلفا گالاکتوزیداز و کلونینگ آن در باکتری E.coli انجام میگیرد. از باکتری E.coli به دلیل دارا بـودن خصوصیاتی مانند سهولت دستکاری ژنتیکی استفاده شد.
مواد و روشها
1- بهینهسازی و ساخت ژن:
در یک مطالعه تجربی، توالی ژن گالاکتوزیداز مربوط به قهوه سبز از پایگاه داده NCBI تهیه شد. این توالی توسط شرکت بیونیر پس از بهینهسازی از نظر کدونها(codon optimization) ساخت و در وکتور pBHA کلون شد. به علاوه جایگاه برش آنزیمهای محدودکننده Nco1 و BamH1 جهت قرارگیری ژن در وکتور نیز به آن اضافه شد که از این جایگاهها برای برش و خارج کردن ژن از وکتور در مراحل بعدی استفاده میشود. ژن ساخته شده با غلظت µg 5 و به صورت لیوفلیزه دریافت شد.
2- آمادهسازی سلولهای مستعد:
جهت آمادهسازی سلولهای مستعد، باکتری E.coli سویه TOP10 در محیطLB Agar فاقد آمپیسیلین کشت داده شد. سایر مراحل به طور خلاصه عبارت بود از: انتخاب یک کلون پس از 20 ساعت نگهداری در انکوباتور و تلقیح به mL 5 محیطBroth LB بدون آمپیسیلین و کشت آن به مدت یک شب. باکتری کشت شده به مدت 10 دقیقه روی یخ قرار داده شد تا رشد باکتری متوقف شود. سپس به فالکون سرد منتقل و در rpm8000 و 4 درجه سانتیگراد به مدت 5 دقیقه سانتریفیوژ و محیط رویی دور ریخته شد. رسوب باکتری در محلول استریل 1/0 مولار CaCl2 سرد سوسپانسیون شد.
3- ترانسفورماسیون به سلولهای مستعد TOP10 :
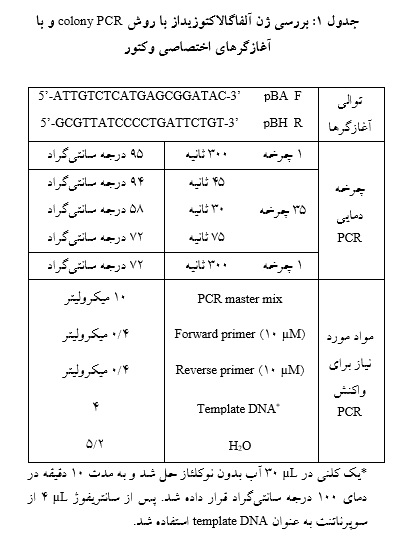
یک میکروتیوب حاوی μL 50 از سلولهای مستعد TOP10 روی یخ قرار داده شد و مقدار μL 2 از پلاسمید pBHA حاوی ژن گالاکتوزیداز ساخته شده با غلظت µg/µL 5/0به میکروتیوب حاوی µL 50 سلولهای مستعد اضافه شد و به مدت 30 دقیقه روی یخ انکوبه شد. مخلوط حاصل به مدت 90 ثانیه در بن ماری 42 درجه سانتیگراد قرار گرفت، سپس فوراً به مدت 5 دقیقه روی یخ قرار داده شد. پس از این مرحله µL 150 محیط کشت LB Broth فاقد آمپیسیلین به میکروتیوب اضافه شد و به مدت 90 دقیقه در 37 درجه سانتیگراد انکوبه شد. مخلوط حاصل به پلیت LB Agar حاوی μg/mL100 آمپیسیلین منتقل، روی سطح آگار گسترده و به مدت یک شب در دمای 37 درجه سانتیگراد انکوبه شد. چند کلنی انتخاب شد و وجود ژن مورد نظر در آنها با روش colony PCR و با آغازگرهای اختصاصی وکتور تائید شد(جدول 1).
4- تکثیر پلاسمید:
پـس از رشـد کلنـیهای حاوی پلاسمید pBHA، یک تک کلون از پلیت انتخاب کرده به mL 20 محیط LB broth حاوی آمپیسیلین(μg/mL100) منتقل گردید. محیط فوق به مدت یک شب در 37 درجه سانتیگراد و شیکر rpm 250 انکوبه شد.
استخراج پلاسمید با استفاده از کیت DENAzist plasmid isolation kit انجام شد.
5- ساب کلون ژن آلفاگالاکتوزیداز در پلاسمید pET-20b :
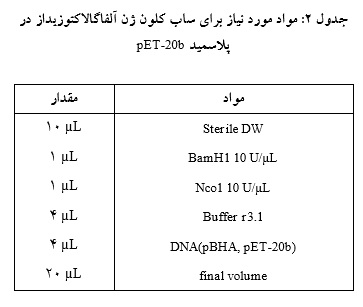
برای بیان ژن آلفا گالاکتوزیداز در E.coli ، قطعه ژنی مورد نظر از پلاسمیدpBHA جداسازی و در پلاسمید بیانی pET-20b ساب کلون شد. برای این منظور، پلاسمید بیانی pET-20b وpBHA حاوی ژن آلفاگالاکتوزیداز در میکروتیوبهای جداگانه با آنزیمهای BamH1 و Nco1 برش خوردند. واکنش هضم آنزیمی پس ازmin 15 انکوباسیون در دمای 37 درجه سانتیگراد و غیرفعال کردن آنزیمها به مدت min 15 در 80 درجه سانتیگراد انجام شد(جدول 2).
6- الکتروفورز قطعات و استخراج از ژل:
جهت جداسازی قطعات حاصل از هضم دو گانه آنزیمی از الکتروفورز قطعات در ژل آگارز 1% استفاده شد. قطعه مورد نظر bp 1475 میباشد. قطعاتی از ژل که حاوی باند مورد نظر بودند در زیر UV برش خوردند و به میکروتیوب استریلmL 5/1 منتقل شدند. جهـت استخراج از ژل از کیت Favor Prep GEL/PCR Purification Kit استفاده شد. پلاسمید بیانی pET-20b با استفاده از آنزیمهای محدود کننده فوق برش داده شد. بعد از اطمینان از خطی شدن پلاسمید با استفاده از ژل آگارز، پلاسمید برش خورده و به منظور جلوگیری از اتصال مجدد دو انتهای آن با آنزیم آلکالین فسفاتاز در انتهای 5 دفسفریله گردید و سپس از روی ژل استخراج و با نانودراپ تعیین غلظت شد.
7- لیگاسیون ژن آلفا گالاکتوزیداز در پلاسمید pET-20b :
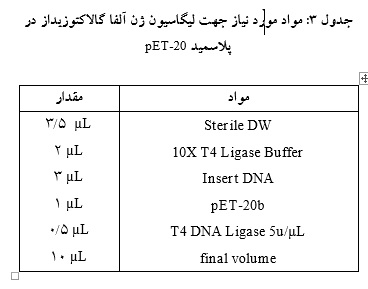
جهت انجام واکنش لیگاسیون، قطعه ژنی و پلاسمید به نسبت 3 به 1 وهمراه آنزیم T4 DNA Ligase با هم ترکیب و به مدت یک شب در دمای محیط نگهداری شد(جدول 3).
8- انتقال پلاسمید به باکتری:
سلولهــای باکتـری TOP10 ترانسفورمـه شـــده روی
محیط LB Agar حاوی آنتیبیوتیک آمپیسیلین رشد داده شدند.
9- غربالگری کلونهای حاوی ژن به روش Colony PCR :
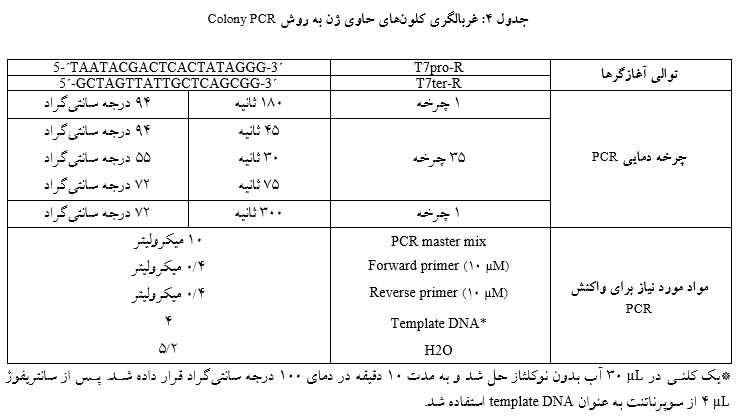
جهت تایید قرار گرفتن ژن آلفاگالاکتوزیداز در پلاسمید از روش Colony PCR استفاده شد. جهت انجام واکنش PCR از آغازگرهای T7Pro-F و T7ter-R و Ampliqon master mix استفاده شد. در صورتی که عمل لایگیشن و ورود قطعات به داخل پلاسمید به نحو درستی انجام شده باشد، طول قطعه حاصل از Colony PCR ، bp 1475 خواهد بود. محصول PCR جهت تعیین توالی ارسال شد. جزئیات برنامه PCR در جدول 4 آورده شده است.
10- تکثیر و جداسازی پلاسمید:
از میان کلنیهایی که وجود ژن در آنها با تعیین توالی تائید شده بود، تعدادی انتخاب و برای تکثیر و استخراج پلاسمید، مورد استفاده قرار گرفت. پلاسمید تکثیر شده در باکتری با استفاده از کیت و طبق دستورالعمل استخراج گردید. پلاسمیدهای استخراج شده پس از تایید انجام شدن کلون توسط دستگاه نانو دراپ تعیین غلظت و بیشترین غلظت از بین نمونهها جهت انجام واکنش ترانسفورمیشن انتخاب گردید.
11- آمادهسازی سلولهای E.coli سویه rosetta 2 (DE3) جهت ترانسفورمیشن:
پلاسمید pET-20b حاوی ژن گالاکتوزیداز با روش شوک حـرارتی وارد باکتری E.coli سویه rosetta 2 (DE3) شد.
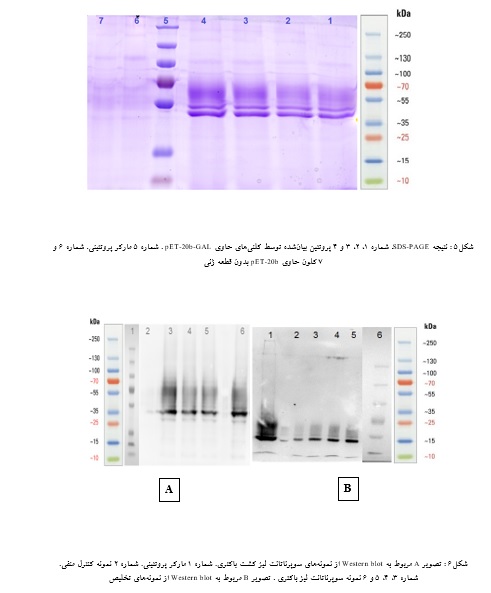
بحث
ابداع سویههای جدید با مهندسی ژنتیک، قدم مهمی در بیان پروتئینهای یوکاریوتی در E.Coli میباشد زیرا این سویهها دارای توانایی اعمال تغییرات پس از ترجمه هستند(11). در این پژوهش ما موفق شدیم آنزیم آلفا گالاکتوزیداز را که توانائی تبدیل گروه خونی B به گروه O را دارد در سویه rosetta 2 (DE3) بیان کنیم. گونههای متداول E.Coli برای بیان پری پلاسمیک پروتئینهای مختلف نوترکیب عبارتند از: BL21 (DE3) ، BL21 (DE3) pLysS ، Rosetta2 (DE3) و Rosetta-gami2 (DE3) آلفا-گالاکتوزیداز از منابع مختلفی(پروانه ابریشم باف، سوسک برگخوار، قهوه سبز، قارچ آسپرژیلوس و.... جـداسـازی شـده و ویـژگیهای آن تعیین شده است. گونههای Rosetta از طریق افزایش کارآیی ترجمه، بیان پروتئینهای هدف را افزایش میدهند(15-11). اگر چه در مطالعه حاضر آنزیم به دست آمده قادر به حذف آنتیژن B از سطح گلبول قرمز میباشد اما فعالیت 100% نبوده بلکه شدت آگلوتیناسیون از +4 قبل از اثر آنزیم به کمتر از +1 بعد از اثر آنزیم کاهش مییابد.
دلایل مختلفی میتواند باعث این امر باشد:
اولاً با توجه به تخلیص آنزیم که فقط با ستون Ni-NTA انجـام گـرفت مـیتوان انتظـار داشــت که غلظـت آنزیـم
در واحد حجم ناچیز بوده و به علاوه وجود مواد ممانعتی(inhibitor) همراه آنزیم موجب کاهش فعالیت آنزیم شده باشد. خالصسازی با روشهای دیگر مانند کروماتوگرافی علاوه بر افزایش غلظت آنزیم میتواند مواد ممانعتی را نیز حذف کرده و باعث ارتقای فعالیت آنزیم شود.
دوماً فعالیت حداکثری هر آنزیم مستلزم استفاده از بافر با pH و غلظت مناسب میباشد که در تحقیق ما به دلیل ضیق وقت انجام نشد.
ثالثاً بهینهسازی شرایط بیان شامل بهترین دما و نیز غلظت مناسب القاکننده(IPTG) باعث خواهد شد که شرایط لازم برای بیان حداکثری مشخص شده و بدینوسیله غلظت آنزیم افزایش یابد. به علاوه تغلیظ آنزیم خالص شده با روشهایی مانند دیالیز به افزایش فعالیت آنزیم کمک خواهد کرد.
یکی از مشکلات بیان آنزیم آلفا گالاکتوزیداز در E.coli ، ایجاد inclusion body بود که فاقد خاصیت آنزیمی بودند. استفاده از سویه مهندسی شده این مشکل را مرتفع ساخته است. برای مثال در مطالعهای که توسط Ahmed و همکاران انجام گرفت، بیان پریپلاسمیک اینترلوکین 15 نوترکیب (rhIL-15) در چهار سویه E.coli بررسی شد. نتایج نشان داد که کمترین بیان rhIL-15 در BL21 (DE3) pLysS وجود دارد در حالی که بیشترین بیان در Rosetta-gami2 (DE3) مشاهده شد. دو سویه دیگر شامل Rosetta2 (DE3) و BL21 (DE3) بیان متوسطی از rhIL-15 نشان دادند(16).
در مطالعههای دیگری نیز از سویههای مختلف برای بیان آنزیم آلفاگالاکتوزیداز استفاده شده است. به عنوان مثال، در مطالعه ژانگ و همکارانش، با کلون کردن آنزیم آلفا گالاکتوزیداز در باکتری و تولید آن توانستند گروه Bرا به گروه O تبدیل کنند. RBCهایی که به این ترتیب تغییر گروه داده بودند از نظر طول عمر، تمامیت غشا، تغییر شکل و مورفولوژی کاملاً شبیه با RBCهای کنترل بودند(17).
در مطالعه ژو و همکارانش، ژن آنزیم آلفا گالاکتوزیداز دانـه قهـوه را کلـون کــرده و آنزیـم را در مقیاس زیاد در
مخمر P.pastoris تولید کردند.
نتایج نشان داد که سکانس به دست آمده در مقایسه با دیگر کلونهای آلفاگالاکتوزیداز استخراج شده از سایر منابع، بین 52% تا 80% دارای همولوژی است. آنها با استفاده از این آنزیم، گروه Bرا به گروه O تبدیل کردند(13). اگر چه نتایج ژو و همکارانش از نظر کمی و کیفی مطلوب بود لکن مشکلات کشت سلول مخمر در مقایسه با باکتری، میتواند نقطه منفی تحقیقات آنها باشد.
در مطالعه گائو و همکـارانش، توانسـتند بـا اسـتفاده از آنزیمهای ان-اسـتیل گالاکتوزآمینیداز و آلفـا-گالاکتوزیداز در یک بافر مشترک، گروه AB را به گروه O تبدیل کنند. واکنش آنزیمی آنها در دمای 4 درجـه سانتیگراد انجام شد. بافرهای مختلفی برای تبدیل آنزیمی گروههای A وB به گروه O آزمایش شد. آنها بهترین نتایج را با بافرهای گلیسین و 5% گلوکز به دست آوردند. از آن جا که گلوکز یکی از اجزای اصلی بافر موجود در کیسه خون میباشد بنابراین، این یافته میتواند پیشرفت بزرگی در معمول شدن تبدیل آنزیمی گروه های خونی باشد(18).
نتیجهگیری
بیان پروتئین یوکاریوتی در میزبان پروکاریوتی میتواند در تولید انبوه آنزیم گالاکتوزیداز برای کاربرد در مراکز انتقال خون کارگشا باشد. بهینهسازی شرایط مختلف بیان برای دستیابی به مقادیر کافی از آنزیم و نیز بهبود شرایط دمایی و شیمیایی واکنش آنزیمی ضروری میباشد.
تشکر و قدردانی
این پژوهش با کد اخلاق IR.IAU.TNB.REC.1400.095 در کمیته اخلاق دانشگاه آزاد اسلامی تهران شمال مورد تصویب قرار گرفته است.
متن کامل: (1744 مشاهده)
کلونینگ آنزیم آلفا گالاکتوزیداز با هدف تبدیل آنتیژن B گروه خونی به آنتیژن O
افسانه حداد دیلمی1، میترا صالحی2، مجید شهابی3
چکیده
سابقه و هدف
کمبود گروههای خونی سیستم ABO یکی از مشکلات متداول مراکز انتقال خون در دنیا میباشد. هم زمان با پیشرفت علم مهندسی ژنتیک و تخلیص پروتئینها، تبدیل آنتیژنهای A و B به آنتیژن O و ایجاد گروه خونی واحد مطرح شد. هدف از این پژوهش، بیان ژن آلفا گالاکتوزیداز در میزبان پروکاریوتی E.coli با ایجاد تغییرات پس از ترجمه و در نهایت تبدیل آنتیژن B گروه خونی به آنتیژن O بود.
مواد و روشها
در این مطالعه تجربی، ژن آلفا گالاکتوزیداز دانه قهوه که پس از بهینهسازی کدونها در پلاسمید pBHA حاوی جایگاه برش آنزیمهای BamH1 و Nco1 کلون شده بود، در باکتری E.coli سویه TOP10 تکثیر و استخراج شد. ژن آلفا گالاکتوزیداز پس از برش با آنزیمهای فوق و تخلیص روی ژل آگاروز مجدداً در پلاسمید بیانی pET-20b کلون شده و به باکتری E.coli سویه rosetta 2 (DE3) وارد شد. بیان ژن با استفاده از القاکننده IPTG انجام شد. وجود پروتئین تولیدی با استفاده از SDS-PAGE و وسترن بلات بررسی شد. آزمون عملکرد پروتئین نیز با توانایی تبدیل گلبولهای B به O ارزیابی شد.
یافتهها
وسترن بلات و SDS-PAGE وجود پروتئین در پری پلاسم باکتری را تائید کرد. آنزیم تخلیص شده توانست آنتیژن B را به طور نسبی از گلبول قرمز حذف کرده و میزان آگلوتیناسیون با آنتیسرم B را به میزان زیادی کاهش دهد.
نتیجه گیری
بیان پروتئین یوکاریوتی در میزبان پروکاریوتی میتواند در تولید انبوه آنزیم گالاکتوزیداز برای کاربرد در مراکز انتقال خون کارگشا باشد. بهینهسازی شرایط مختلف بیان برای دستیابی به مقادیر کافی از آنزیم ضروری میباشد.
کلمات کلیدی: گروه خونی، گالاکتوزیداز، E.coli
تاریخ دریافت: 08/03/1401
تاریخ پذیرش: 19/06/1401
1- دانشجوی کارشناسی ارشد بیوتکنولوژی ـ دانشکده علوم زیستی دانشگاه آزاد اسلامی واحد تهران شمال ـ تهران ـ ایران
2- مؤلف مسئول: دکترای تخصصی بیولوژی سلولی و مولکولی با گرایش باکتریولوژی مولکولیـ استادیار دانشکده علوم زیستی دانشگاه آزاد اسلامی واحد تهران شمال ـ تهران ـ ایران ـ کد پستی: 1651153511
3- PhD فرآوردههای بیولوژیک ـ استادیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
افسانه حداد دیلمی1، میترا صالحی2، مجید شهابی3
چکیده
سابقه و هدف
کمبود گروههای خونی سیستم ABO یکی از مشکلات متداول مراکز انتقال خون در دنیا میباشد. هم زمان با پیشرفت علم مهندسی ژنتیک و تخلیص پروتئینها، تبدیل آنتیژنهای A و B به آنتیژن O و ایجاد گروه خونی واحد مطرح شد. هدف از این پژوهش، بیان ژن آلفا گالاکتوزیداز در میزبان پروکاریوتی E.coli با ایجاد تغییرات پس از ترجمه و در نهایت تبدیل آنتیژن B گروه خونی به آنتیژن O بود.
مواد و روشها
در این مطالعه تجربی، ژن آلفا گالاکتوزیداز دانه قهوه که پس از بهینهسازی کدونها در پلاسمید pBHA حاوی جایگاه برش آنزیمهای BamH1 و Nco1 کلون شده بود، در باکتری E.coli سویه TOP10 تکثیر و استخراج شد. ژن آلفا گالاکتوزیداز پس از برش با آنزیمهای فوق و تخلیص روی ژل آگاروز مجدداً در پلاسمید بیانی pET-20b کلون شده و به باکتری E.coli سویه rosetta 2 (DE3) وارد شد. بیان ژن با استفاده از القاکننده IPTG انجام شد. وجود پروتئین تولیدی با استفاده از SDS-PAGE و وسترن بلات بررسی شد. آزمون عملکرد پروتئین نیز با توانایی تبدیل گلبولهای B به O ارزیابی شد.
یافتهها
وسترن بلات و SDS-PAGE وجود پروتئین در پری پلاسم باکتری را تائید کرد. آنزیم تخلیص شده توانست آنتیژن B را به طور نسبی از گلبول قرمز حذف کرده و میزان آگلوتیناسیون با آنتیسرم B را به میزان زیادی کاهش دهد.
نتیجه گیری
بیان پروتئین یوکاریوتی در میزبان پروکاریوتی میتواند در تولید انبوه آنزیم گالاکتوزیداز برای کاربرد در مراکز انتقال خون کارگشا باشد. بهینهسازی شرایط مختلف بیان برای دستیابی به مقادیر کافی از آنزیم ضروری میباشد.
کلمات کلیدی: گروه خونی، گالاکتوزیداز، E.coli
تاریخ دریافت: 08/03/1401
تاریخ پذیرش: 19/06/1401
1- دانشجوی کارشناسی ارشد بیوتکنولوژی ـ دانشکده علوم زیستی دانشگاه آزاد اسلامی واحد تهران شمال ـ تهران ـ ایران
2- مؤلف مسئول: دکترای تخصصی بیولوژی سلولی و مولکولی با گرایش باکتریولوژی مولکولیـ استادیار دانشکده علوم زیستی دانشگاه آزاد اسلامی واحد تهران شمال ـ تهران ـ ایران ـ کد پستی: 1651153511
3- PhD فرآوردههای بیولوژیک ـ استادیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
مقدمه
سیستم گروه خونی ABO مهمترین سیستم گروه خونی در طب انتقال خون میباشد. پایه آنتیژنهای این سیستم ماده H میباشد که در سطح تمام گلبولهای قرمز وجود دارد. جایگاه ژنی ABO داری سه آلل A ، B و O میباشد(1).
آللهای A و B آنزیمهای ترانسفرازی را کد میکنند که باعث اتصال واحدهای کربوهیدراتی به ماده H میشوند. محصول پروتئینی آلل A آنزیم ان استیل گالاکتوزآمین ترانسفراز است که یک مولکول ان استیل گالاکتوزآمین را به ماده H متصل میکند و آنتیژن A را به وجود می آورد. آلل Bآنزیم گالاکتوزیل ترانسفراز را کد میکند که با افزودن یک مولکول گالاکتوز به ماده Hآن را تبدیل به آنتیژن B میکند. آلل O فاقد محصول خاصی بوده و افراد هموزیگوت برای این آلل، ماده H را به صورت دست نخورده در سطح گلبولهای قرمز خود بیان میکنند.
به دلیل وجود آنتیبادیهای ABO در همه افراد، بدون مواجهه قبلی با گلبولهای قرمز انسانی، گروهبندی صحیح اهداکننده و دریافتکننده خون از نظر ABO ، اساس ایمنی انتقال خون را تشکیل میدهد. تزریق خون ناسازگار از نظر ABO به بیمار میتواند منجر به همولیز داخل عروقی و عوارض جدی ناشی از واکنش انتقال خون حاد شود(3، 2).
در بین گروههای خونی، گروه O به عنوان دهنده همگانی برای بسیاری از بیماران به ویژه در موارد اورژانسی که امکان تعیین گروه ABO میسر نیست مورد استفاده قرار میگیرد(4). بررسی فراوانی گروههای ABO در جمعیت ایرانی نشان داده که گروه O و A بیشترین درصد جمعیت را تشکیل میدهند(به ترتیب 49/36 و 09/32 درصد). به دنبال آنها گروه B با فراوانی 68/23% و گروه AB با فراوانی 74/7% قرار دارند(5).
اندیشه تبدیل همه گروههای سیستم ABOبه گروه O از ابتدای دهه 1990میلادی مطرح شد. در بین گروههای خونی، آنتیژن B فقط در گالاکتوز انتهایی با آنتیژن O متفاوت است. بنابراین با استفاده از آنزیم گالاکتوزیداز (α-galactosidas)، میتوان گروه B را به O تبدیل کرد(8-6). از آن جا که ناخالصیهای همراه با آنزیم استخراج شده از منابع گیاهی میتوانست منجر به بروز عوارض و واکنشهای ناخواسته شود. از این رو تکنولوژی DNA نوترکیب مورد استفاده گرفت. استفاده از این تکنولوژی، امکان تولید آنزیم را در مقیاس وسیع و با آلودگی کمتر امکانپذیر ساخته است. به علاوه، مشخص شده که نوع نوترکیب در مقایسه با آلفاگالاکتوزیداز استخراج شده از منابع گیاهی(دانه سبز قهوه) بسیار کارآمدتر است(10، 9).
در صورتی که امکان تبدیل خون افراد گروه B به گروه O میسر شود، گام مهمی در جهت فراهم آوردن ذخیره خونی میباشد. یکی از مشکلات در این زمینه تولید انبوه آنزیم آلفاگالاکتوزیداز با کارآیی مناسب و در مقیاس وسیع است.
لذا مطالعه حاضر، با هدف تولید آنزیم بهینهسازی شده آلفا گالاکتوزیداز و کلونینگ آن در باکتری E.coli انجام میگیرد. از باکتری E.coli به دلیل دارا بـودن خصوصیاتی مانند سهولت دستکاری ژنتیکی استفاده شد.
مواد و روشها
1- بهینهسازی و ساخت ژن:
در یک مطالعه تجربی، توالی ژن گالاکتوزیداز مربوط به قهوه سبز از پایگاه داده NCBI تهیه شد. این توالی توسط شرکت بیونیر پس از بهینهسازی از نظر کدونها(codon optimization) ساخت و در وکتور pBHA کلون شد. به علاوه جایگاه برش آنزیمهای محدودکننده Nco1 و BamH1 جهت قرارگیری ژن در وکتور نیز به آن اضافه شد که از این جایگاهها برای برش و خارج کردن ژن از وکتور در مراحل بعدی استفاده میشود. ژن ساخته شده با غلظت µg 5 و به صورت لیوفلیزه دریافت شد.
2- آمادهسازی سلولهای مستعد:
جهت آمادهسازی سلولهای مستعد، باکتری E.coli سویه TOP10 در محیطLB Agar فاقد آمپیسیلین کشت داده شد. سایر مراحل به طور خلاصه عبارت بود از: انتخاب یک کلون پس از 20 ساعت نگهداری در انکوباتور و تلقیح به mL 5 محیطBroth LB بدون آمپیسیلین و کشت آن به مدت یک شب. باکتری کشت شده به مدت 10 دقیقه روی یخ قرار داده شد تا رشد باکتری متوقف شود. سپس به فالکون سرد منتقل و در rpm8000 و 4 درجه سانتیگراد به مدت 5 دقیقه سانتریفیوژ و محیط رویی دور ریخته شد. رسوب باکتری در محلول استریل 1/0 مولار CaCl2 سرد سوسپانسیون شد.
3- ترانسفورماسیون به سلولهای مستعد TOP10 :
یک میکروتیوب حاوی μL 50 از سلولهای مستعد TOP10 روی یخ قرار داده شد و مقدار μL 2 از پلاسمید pBHA حاوی ژن گالاکتوزیداز ساخته شده با غلظت µg/µL 5/0به میکروتیوب حاوی µL 50 سلولهای مستعد اضافه شد و به مدت 30 دقیقه روی یخ انکوبه شد. مخلوط حاصل به مدت 90 ثانیه در بن ماری 42 درجه سانتیگراد قرار گرفت، سپس فوراً به مدت 5 دقیقه روی یخ قرار داده شد. پس از این مرحله µL 150 محیط کشت LB Broth فاقد آمپیسیلین به میکروتیوب اضافه شد و به مدت 90 دقیقه در 37 درجه سانتیگراد انکوبه شد. مخلوط حاصل به پلیت LB Agar حاوی μg/mL100 آمپیسیلین منتقل، روی سطح آگار گسترده و به مدت یک شب در دمای 37 درجه سانتیگراد انکوبه شد. چند کلنی انتخاب شد و وجود ژن مورد نظر در آنها با روش colony PCR و با آغازگرهای اختصاصی وکتور تائید شد(جدول 1).
4- تکثیر پلاسمید:
پـس از رشـد کلنـیهای حاوی پلاسمید pBHA، یک تک کلون از پلیت انتخاب کرده به mL 20 محیط LB broth حاوی آمپیسیلین(μg/mL100) منتقل گردید. محیط فوق به مدت یک شب در 37 درجه سانتیگراد و شیکر rpm 250 انکوبه شد.
استخراج پلاسمید با استفاده از کیت DENAzist plasmid isolation kit انجام شد.
5- ساب کلون ژن آلفاگالاکتوزیداز در پلاسمید pET-20b :
برای بیان ژن آلفا گالاکتوزیداز در E.coli ، قطعه ژنی مورد نظر از پلاسمیدpBHA جداسازی و در پلاسمید بیانی pET-20b ساب کلون شد. برای این منظور، پلاسمید بیانی pET-20b وpBHA حاوی ژن آلفاگالاکتوزیداز در میکروتیوبهای جداگانه با آنزیمهای BamH1 و Nco1 برش خوردند. واکنش هضم آنزیمی پس ازmin 15 انکوباسیون در دمای 37 درجه سانتیگراد و غیرفعال کردن آنزیمها به مدت min 15 در 80 درجه سانتیگراد انجام شد(جدول 2).
6- الکتروفورز قطعات و استخراج از ژل:
جهت جداسازی قطعات حاصل از هضم دو گانه آنزیمی از الکتروفورز قطعات در ژل آگارز 1% استفاده شد. قطعه مورد نظر bp 1475 میباشد. قطعاتی از ژل که حاوی باند مورد نظر بودند در زیر UV برش خوردند و به میکروتیوب استریلmL 5/1 منتقل شدند. جهـت استخراج از ژل از کیت Favor Prep GEL/PCR Purification Kit استفاده شد. پلاسمید بیانی pET-20b با استفاده از آنزیمهای محدود کننده فوق برش داده شد. بعد از اطمینان از خطی شدن پلاسمید با استفاده از ژل آگارز، پلاسمید برش خورده و به منظور جلوگیری از اتصال مجدد دو انتهای آن با آنزیم آلکالین فسفاتاز در انتهای 5 دفسفریله گردید و سپس از روی ژل استخراج و با نانودراپ تعیین غلظت شد.
7- لیگاسیون ژن آلفا گالاکتوزیداز در پلاسمید pET-20b :
جهت انجام واکنش لیگاسیون، قطعه ژنی و پلاسمید به نسبت 3 به 1 وهمراه آنزیم T4 DNA Ligase با هم ترکیب و به مدت یک شب در دمای محیط نگهداری شد(جدول 3).
8- انتقال پلاسمید به باکتری:
سلولهــای باکتـری TOP10 ترانسفورمـه شـــده روی
محیط LB Agar حاوی آنتیبیوتیک آمپیسیلین رشد داده شدند.
9- غربالگری کلونهای حاوی ژن به روش Colony PCR :
جهت تایید قرار گرفتن ژن آلفاگالاکتوزیداز در پلاسمید از روش Colony PCR استفاده شد. جهت انجام واکنش PCR از آغازگرهای T7Pro-F و T7ter-R و Ampliqon master mix استفاده شد. در صورتی که عمل لایگیشن و ورود قطعات به داخل پلاسمید به نحو درستی انجام شده باشد، طول قطعه حاصل از Colony PCR ، bp 1475 خواهد بود. محصول PCR جهت تعیین توالی ارسال شد. جزئیات برنامه PCR در جدول 4 آورده شده است.
10- تکثیر و جداسازی پلاسمید:
از میان کلنیهایی که وجود ژن در آنها با تعیین توالی تائید شده بود، تعدادی انتخاب و برای تکثیر و استخراج پلاسمید، مورد استفاده قرار گرفت. پلاسمید تکثیر شده در باکتری با استفاده از کیت و طبق دستورالعمل استخراج گردید. پلاسمیدهای استخراج شده پس از تایید انجام شدن کلون توسط دستگاه نانو دراپ تعیین غلظت و بیشترین غلظت از بین نمونهها جهت انجام واکنش ترانسفورمیشن انتخاب گردید.
11- آمادهسازی سلولهای E.coli سویه rosetta 2 (DE3) جهت ترانسفورمیشن:
پلاسمید pET-20b حاوی ژن گالاکتوزیداز با روش شوک حـرارتی وارد باکتری E.coli سویه rosetta 2 (DE3) شد.
باکتری ترانسفورم شده روی محیط LB آگار، حاوی آنتیبیوتیکهای آمپیسیلین(µg/mL 50) کشت داده شد و به صورت شبانه در دمای 37 درجه سانتیگراد نگهداری شد. سپس چهار کلنی انتخاب و روی محیط مایع LB حاوی آمپیسیلین به صورت شبانه در دمای 37 درجه سانتیگراد نگهداری شد. در مرحله بعد 1 میلیلیتر از کشت فوق با 100 میلیلیتر محیط کشت تازه حاوی آنتیبیوتیک مخلوط شده و در دمای 37 درجه سانتیگراد نگهداری شد تا جذب نوری محیط(OD600) به محدوده 1-6/0 برسد. در این مرحله محلول IPTG با غلظت 1/0 مولار اضافه شده و در دمای 37 درجه سانتیگراد به صورت شبانه انکوبه شد.
12- خالصسازی آنزیم ترشحی در محیط کشت:
محیط کشت در دور 14000 و به مدت 10 دقیقه سانتریفوژ شد. پلیت باکتریایی در 10 میلیلیتر محلول PBS سوسپانسیون شد. جدار باکتریها با امواج اولتراسوند (sonication) روی یخ پاره شد. سپس محلول به دست آمده در دمای 4 درجه سانتیگراد با دور 14000 و به مدت 3 دقیقه سانتریفوژ شد. هر دو جزء حاصل (سوپرناتنت و پلیت) با SDS-PAGE با غلظت 15% الکتروفورز شده و با رنگ کومازینبلو رنگآمیزی و بررسی شدند. با توجه به وجود پروتئین نوترکیب در پریپلاسم، و نیز دارا بودن توالی His-tag در پروتئین، سوپرناتنت حاصل از لیز باکتریها با کمک ستون تمایلی نیکل(Ni-NTA) تخلیص صورت گرفت. برای اطمینان از تخلیص پروتئین مورد نظر پروتئین تخلیص شده ابتدا با SDS-PAGE الکتروفورز شده و با آنتیبادی ضد His tag وسترن بلات انجام شد.
13- وسترن بلات:
به منظور تایید نهایی نمونههایی که SDS-PAGE در آنها دارای باند مورد نظر میباشد، وسترن بلات انجام شد. پس از الکتروفورز پروتئینها، انتقال باندهای پروتئین جدا شده از ژل پلیآکریل آمید به کاغذ نیترو سلولز به وسیله جریان الکتریسیته(mA 300 به مدت 60) دقیقه انجام شد. کاغذ به مدت 180 دقیقه در 10 میلیلیتر محلول بلاکر در 4 درجه سانتیگراد انکوبه و سپس کاغذ از بلاکر خارج و 3 بار با بافر TBS-T شستشو داده شد. آنتیبادی اولیه (mouse anti-histag) با محلول بلاکر به نسبت 1:1000 رقیق و به کاغذ نیتروسلولز اضافه شد و به صورت شبانه در 4 درجه سانتیگراد انکوبه شد. کاغذ از محلول خارج و سه بار با TBS-T شستشو داده شد. در مرحله بعد کاغذ با آنتی بادی ثانویه ((Anti-mouse IgG-HRP به مدت 90 دقیقه در 4 درجه سانتیگراد انکوبه شد. کاغذ از محلول خارج و سه بار با TBS-T شستشو داده شد و سپس با محلول ECL (Enhanced chemiluminescence) به مدت 1 دقیقه انکوبه گردید. کاغذ در تاریکی نگه داشته شد و با دستگاه کمی لومینسنس بررسی شد.
14- تخلیص آنزیم:
به دلیل وجود توالی His tag تخلیص با کمک ستون تمایلی نیکل Ni-NTA صورت گرفت.
15- آزمایش اولیه فعالیت آنزیم:
به منظور سنجش فعالیت آنزیم، آزمون اولیه تاثیر آنزیم بر گلبول قرمز انجام شد. بدین منظور از گلبولهای گروه B سوسپانسیون 5% در سالین تهیه شد. میکروتیوب حاوی 100 میکرولیتر گلبول قرمز %5 در سالین با مقادیر متفاوت از آنزیم تخلیص شده (µL 1000 و 750 ، 500 ، 250) در دمای اتاق به مدت 2 ساعت مجاور گردید. سپس به گلبولها آنتیسرم B اضافه و سانتریفوژ شد. آزمون کراسمچ نیز با استفاده از ایـن گلبولهـا و سـرم افـراد بـا گروههای مختلف انجام شد.
یافتهها
نتیجه clony PCR :
پس از ترانسفورم کردن باکتری TOP10 با پلاسمید pBHA حاوی ژن گالاکتوزیداز و کشت باکتری به صورت شبانه، colony PCR انجام شد (شکل 1).
استخراج پلاسمید:
پـس از تاییـد حـضــور ژن GAL در پـلاسمیــدهـای
pBHA و در مراحل بعد در pET-20b حاصل از لیگاسیون، از آنها برای تکثیر و استخراج پلاسمید استفاده شد. شکل 2 نشاندهنده الکتروفورز این پلاسمیدها روی آگاروز 1% میباشد.

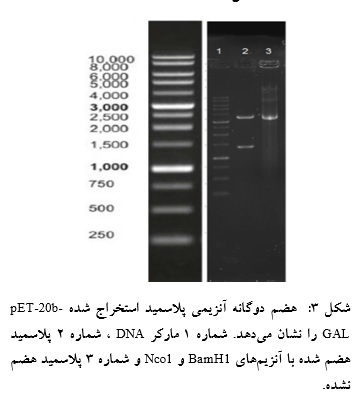
تاییـد پلاسمید با هضم دوگانه آنزیمی:
پس از استخراج پلاسمید برای تایید نهایی و قبل از ترانسفورمه کردن باکتری rosetta 2 ، پلاسمید استخراج شده با استفاده از آنزیمهای BamH1 و Nco1 هضم آنزیمی
گردید. الکتروفورز محصولات هضم آنزیمی در شکل نشان
داده شده است(شکل 3).
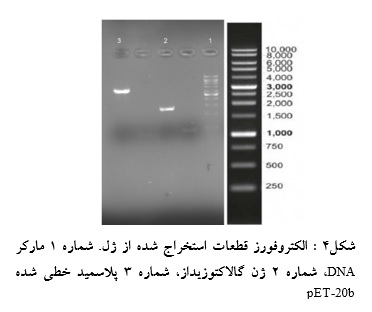
استخراج از ژل:
قطعات حاصل از هضم دوگانه آنزیمی پلاسمید pET-20b پس از استخراج از ژل بر روی ژل آگارز الکتروفورز گردید تا صحت استخراج از ژل و غلظت باندهای حاصل بررسی شود (شکل 4).
تایید اتصال قطعه به روش colony PCR :
پس از انجام لیگاسیون، سویه مستعد TOP10 با مخلوط لیگاسیون ترنسفورم شد. غربالگری جهت تایید ورود ژن GAL به درون پلاسمیدpET-20b با استفاده از روش colony PCR انجام شد.تکثیر قطعه GAL با آغازگرهای T7pro-F و T7ter-R انجام شد که تکثیر این قطعه با این آغازگرها منجر به ایجاد قطعهای با طول bp 1475شد(شکل 5).
نتیجه وسترن بلات:
با توجه به وجود His-tag در انتهای N پروتئین بیان شده، وسترن بلات با استفاده از آنتیبادی اولیه (C-myc Mouse monoclonal IgG1) و آنتیبادی ثانویه(Anti-Mouse IgG-HRP) انجام شد. باندهای روشن در ناحیه KDa 55 نشاندهنده پروتئین مورد نظر میباشد(شکل6). پروتئین نوترکیب بیان شده علاوه بر His Tag دارای توالی c-myc هم بوده است لذا چون آنتیبادی ضد c-myc موجود بود، از آن استفاده شد. ضمناً در پروتئین محل قرار گرفتن c-myc در انتهای کربوکسی ترمینال و قبل از His Tag میباشد.
لذا چنانچه پروتئین به طور کامل ترجمه نشده و فاقد His tag باشد باز هم با آنتیبادی ضد c-myc قابل تشخیص خواهد بود و این مزیت استفاده از آنتیبادی c-myc است. از His tag برای تخلیص استفاده شد.
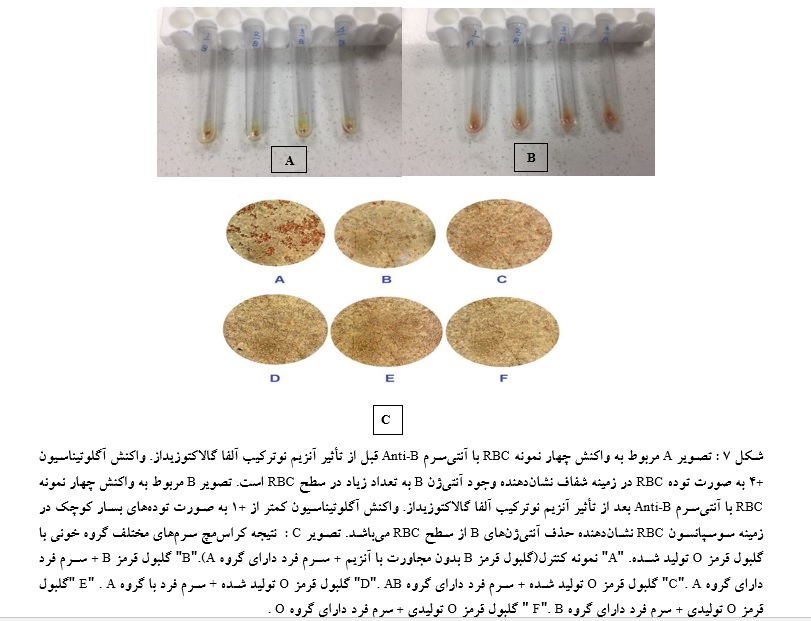
بررسی فعالیت آنزیم:
گلبولهای قرمز شسته شده در مجاورت آنزیم قرار گرفته و سپس توسط آنتیسرم B از نظر وجود آنتیژن B مورد ارزیابی قرار گرفتند. آزمون کراسمچ نمونه گروه خونی O تولید شده با سرمهای گروههای خونی A و B و AB و O انجام شد و نتیجه با میکروسکوپ بررسی شد. دو نمونه به عنوان کنترل و 4 نمونه جهت آزمایش بررسی شدند(شکل 7).
12- خالصسازی آنزیم ترشحی در محیط کشت:
محیط کشت در دور 14000 و به مدت 10 دقیقه سانتریفوژ شد. پلیت باکتریایی در 10 میلیلیتر محلول PBS سوسپانسیون شد. جدار باکتریها با امواج اولتراسوند (sonication) روی یخ پاره شد. سپس محلول به دست آمده در دمای 4 درجه سانتیگراد با دور 14000 و به مدت 3 دقیقه سانتریفوژ شد. هر دو جزء حاصل (سوپرناتنت و پلیت) با SDS-PAGE با غلظت 15% الکتروفورز شده و با رنگ کومازینبلو رنگآمیزی و بررسی شدند. با توجه به وجود پروتئین نوترکیب در پریپلاسم، و نیز دارا بودن توالی His-tag در پروتئین، سوپرناتنت حاصل از لیز باکتریها با کمک ستون تمایلی نیکل(Ni-NTA) تخلیص صورت گرفت. برای اطمینان از تخلیص پروتئین مورد نظر پروتئین تخلیص شده ابتدا با SDS-PAGE الکتروفورز شده و با آنتیبادی ضد His tag وسترن بلات انجام شد.
13- وسترن بلات:
به منظور تایید نهایی نمونههایی که SDS-PAGE در آنها دارای باند مورد نظر میباشد، وسترن بلات انجام شد. پس از الکتروفورز پروتئینها، انتقال باندهای پروتئین جدا شده از ژل پلیآکریل آمید به کاغذ نیترو سلولز به وسیله جریان الکتریسیته(mA 300 به مدت 60) دقیقه انجام شد. کاغذ به مدت 180 دقیقه در 10 میلیلیتر محلول بلاکر در 4 درجه سانتیگراد انکوبه و سپس کاغذ از بلاکر خارج و 3 بار با بافر TBS-T شستشو داده شد. آنتیبادی اولیه (mouse anti-histag) با محلول بلاکر به نسبت 1:1000 رقیق و به کاغذ نیتروسلولز اضافه شد و به صورت شبانه در 4 درجه سانتیگراد انکوبه شد. کاغذ از محلول خارج و سه بار با TBS-T شستشو داده شد. در مرحله بعد کاغذ با آنتی بادی ثانویه ((Anti-mouse IgG-HRP به مدت 90 دقیقه در 4 درجه سانتیگراد انکوبه شد. کاغذ از محلول خارج و سه بار با TBS-T شستشو داده شد و سپس با محلول ECL (Enhanced chemiluminescence) به مدت 1 دقیقه انکوبه گردید. کاغذ در تاریکی نگه داشته شد و با دستگاه کمی لومینسنس بررسی شد.
14- تخلیص آنزیم:
به دلیل وجود توالی His tag تخلیص با کمک ستون تمایلی نیکل Ni-NTA صورت گرفت.
15- آزمایش اولیه فعالیت آنزیم:
به منظور سنجش فعالیت آنزیم، آزمون اولیه تاثیر آنزیم بر گلبول قرمز انجام شد. بدین منظور از گلبولهای گروه B سوسپانسیون 5% در سالین تهیه شد. میکروتیوب حاوی 100 میکرولیتر گلبول قرمز %5 در سالین با مقادیر متفاوت از آنزیم تخلیص شده (µL 1000 و 750 ، 500 ، 250) در دمای اتاق به مدت 2 ساعت مجاور گردید. سپس به گلبولها آنتیسرم B اضافه و سانتریفوژ شد. آزمون کراسمچ نیز با استفاده از ایـن گلبولهـا و سـرم افـراد بـا گروههای مختلف انجام شد.
یافتهها
نتیجه clony PCR :
پس از ترانسفورم کردن باکتری TOP10 با پلاسمید pBHA حاوی ژن گالاکتوزیداز و کشت باکتری به صورت شبانه، colony PCR انجام شد (شکل 1).
استخراج پلاسمید:
پـس از تاییـد حـضــور ژن GAL در پـلاسمیــدهـای
pBHA و در مراحل بعد در pET-20b حاصل از لیگاسیون، از آنها برای تکثیر و استخراج پلاسمید استفاده شد. شکل 2 نشاندهنده الکتروفورز این پلاسمیدها روی آگاروز 1% میباشد.
تاییـد پلاسمید با هضم دوگانه آنزیمی:
پس از استخراج پلاسمید برای تایید نهایی و قبل از ترانسفورمه کردن باکتری rosetta 2 ، پلاسمید استخراج شده با استفاده از آنزیمهای BamH1 و Nco1 هضم آنزیمی
گردید. الکتروفورز محصولات هضم آنزیمی در شکل نشان
داده شده است(شکل 3).
استخراج از ژل:
قطعات حاصل از هضم دوگانه آنزیمی پلاسمید pET-20b پس از استخراج از ژل بر روی ژل آگارز الکتروفورز گردید تا صحت استخراج از ژل و غلظت باندهای حاصل بررسی شود (شکل 4).
تایید اتصال قطعه به روش colony PCR :
پس از انجام لیگاسیون، سویه مستعد TOP10 با مخلوط لیگاسیون ترنسفورم شد. غربالگری جهت تایید ورود ژن GAL به درون پلاسمیدpET-20b با استفاده از روش colony PCR انجام شد.تکثیر قطعه GAL با آغازگرهای T7pro-F و T7ter-R انجام شد که تکثیر این قطعه با این آغازگرها منجر به ایجاد قطعهای با طول bp 1475شد(شکل 5).
نتیجه وسترن بلات:
با توجه به وجود His-tag در انتهای N پروتئین بیان شده، وسترن بلات با استفاده از آنتیبادی اولیه (C-myc Mouse monoclonal IgG1) و آنتیبادی ثانویه(Anti-Mouse IgG-HRP) انجام شد. باندهای روشن در ناحیه KDa 55 نشاندهنده پروتئین مورد نظر میباشد(شکل6). پروتئین نوترکیب بیان شده علاوه بر His Tag دارای توالی c-myc هم بوده است لذا چون آنتیبادی ضد c-myc موجود بود، از آن استفاده شد. ضمناً در پروتئین محل قرار گرفتن c-myc در انتهای کربوکسی ترمینال و قبل از His Tag میباشد.
لذا چنانچه پروتئین به طور کامل ترجمه نشده و فاقد His tag باشد باز هم با آنتیبادی ضد c-myc قابل تشخیص خواهد بود و این مزیت استفاده از آنتیبادی c-myc است. از His tag برای تخلیص استفاده شد.
بررسی فعالیت آنزیم:
گلبولهای قرمز شسته شده در مجاورت آنزیم قرار گرفته و سپس توسط آنتیسرم B از نظر وجود آنتیژن B مورد ارزیابی قرار گرفتند. آزمون کراسمچ نمونه گروه خونی O تولید شده با سرمهای گروههای خونی A و B و AB و O انجام شد و نتیجه با میکروسکوپ بررسی شد. دو نمونه به عنوان کنترل و 4 نمونه جهت آزمایش بررسی شدند(شکل 7).
ابداع سویههای جدید با مهندسی ژنتیک، قدم مهمی در بیان پروتئینهای یوکاریوتی در E.Coli میباشد زیرا این سویهها دارای توانایی اعمال تغییرات پس از ترجمه هستند(11). در این پژوهش ما موفق شدیم آنزیم آلفا گالاکتوزیداز را که توانائی تبدیل گروه خونی B به گروه O را دارد در سویه rosetta 2 (DE3) بیان کنیم. گونههای متداول E.Coli برای بیان پری پلاسمیک پروتئینهای مختلف نوترکیب عبارتند از: BL21 (DE3) ، BL21 (DE3) pLysS ، Rosetta2 (DE3) و Rosetta-gami2 (DE3) آلفا-گالاکتوزیداز از منابع مختلفی(پروانه ابریشم باف، سوسک برگخوار، قهوه سبز، قارچ آسپرژیلوس و.... جـداسـازی شـده و ویـژگیهای آن تعیین شده است. گونههای Rosetta از طریق افزایش کارآیی ترجمه، بیان پروتئینهای هدف را افزایش میدهند(15-11). اگر چه در مطالعه حاضر آنزیم به دست آمده قادر به حذف آنتیژن B از سطح گلبول قرمز میباشد اما فعالیت 100% نبوده بلکه شدت آگلوتیناسیون از +4 قبل از اثر آنزیم به کمتر از +1 بعد از اثر آنزیم کاهش مییابد.
دلایل مختلفی میتواند باعث این امر باشد:
اولاً با توجه به تخلیص آنزیم که فقط با ستون Ni-NTA انجـام گـرفت مـیتوان انتظـار داشــت که غلظـت آنزیـم
در واحد حجم ناچیز بوده و به علاوه وجود مواد ممانعتی(inhibitor) همراه آنزیم موجب کاهش فعالیت آنزیم شده باشد. خالصسازی با روشهای دیگر مانند کروماتوگرافی علاوه بر افزایش غلظت آنزیم میتواند مواد ممانعتی را نیز حذف کرده و باعث ارتقای فعالیت آنزیم شود.
دوماً فعالیت حداکثری هر آنزیم مستلزم استفاده از بافر با pH و غلظت مناسب میباشد که در تحقیق ما به دلیل ضیق وقت انجام نشد.
ثالثاً بهینهسازی شرایط بیان شامل بهترین دما و نیز غلظت مناسب القاکننده(IPTG) باعث خواهد شد که شرایط لازم برای بیان حداکثری مشخص شده و بدینوسیله غلظت آنزیم افزایش یابد. به علاوه تغلیظ آنزیم خالص شده با روشهایی مانند دیالیز به افزایش فعالیت آنزیم کمک خواهد کرد.
یکی از مشکلات بیان آنزیم آلفا گالاکتوزیداز در E.coli ، ایجاد inclusion body بود که فاقد خاصیت آنزیمی بودند. استفاده از سویه مهندسی شده این مشکل را مرتفع ساخته است. برای مثال در مطالعهای که توسط Ahmed و همکاران انجام گرفت، بیان پریپلاسمیک اینترلوکین 15 نوترکیب (rhIL-15) در چهار سویه E.coli بررسی شد. نتایج نشان داد که کمترین بیان rhIL-15 در BL21 (DE3) pLysS وجود دارد در حالی که بیشترین بیان در Rosetta-gami2 (DE3) مشاهده شد. دو سویه دیگر شامل Rosetta2 (DE3) و BL21 (DE3) بیان متوسطی از rhIL-15 نشان دادند(16).
در مطالعههای دیگری نیز از سویههای مختلف برای بیان آنزیم آلفاگالاکتوزیداز استفاده شده است. به عنوان مثال، در مطالعه ژانگ و همکارانش، با کلون کردن آنزیم آلفا گالاکتوزیداز در باکتری و تولید آن توانستند گروه Bرا به گروه O تبدیل کنند. RBCهایی که به این ترتیب تغییر گروه داده بودند از نظر طول عمر، تمامیت غشا، تغییر شکل و مورفولوژی کاملاً شبیه با RBCهای کنترل بودند(17).
در مطالعه ژو و همکارانش، ژن آنزیم آلفا گالاکتوزیداز دانـه قهـوه را کلـون کــرده و آنزیـم را در مقیاس زیاد در
مخمر P.pastoris تولید کردند.
نتایج نشان داد که سکانس به دست آمده در مقایسه با دیگر کلونهای آلفاگالاکتوزیداز استخراج شده از سایر منابع، بین 52% تا 80% دارای همولوژی است. آنها با استفاده از این آنزیم، گروه Bرا به گروه O تبدیل کردند(13). اگر چه نتایج ژو و همکارانش از نظر کمی و کیفی مطلوب بود لکن مشکلات کشت سلول مخمر در مقایسه با باکتری، میتواند نقطه منفی تحقیقات آنها باشد.
در مطالعه گائو و همکـارانش، توانسـتند بـا اسـتفاده از آنزیمهای ان-اسـتیل گالاکتوزآمینیداز و آلفـا-گالاکتوزیداز در یک بافر مشترک، گروه AB را به گروه O تبدیل کنند. واکنش آنزیمی آنها در دمای 4 درجـه سانتیگراد انجام شد. بافرهای مختلفی برای تبدیل آنزیمی گروههای A وB به گروه O آزمایش شد. آنها بهترین نتایج را با بافرهای گلیسین و 5% گلوکز به دست آوردند. از آن جا که گلوکز یکی از اجزای اصلی بافر موجود در کیسه خون میباشد بنابراین، این یافته میتواند پیشرفت بزرگی در معمول شدن تبدیل آنزیمی گروه های خونی باشد(18).
نتیجهگیری
بیان پروتئین یوکاریوتی در میزبان پروکاریوتی میتواند در تولید انبوه آنزیم گالاکتوزیداز برای کاربرد در مراکز انتقال خون کارگشا باشد. بهینهسازی شرایط مختلف بیان برای دستیابی به مقادیر کافی از آنزیم و نیز بهبود شرایط دمایی و شیمیایی واکنش آنزیمی ضروری میباشد.
تشکر و قدردانی
این پژوهش با کد اخلاق IR.IAU.TNB.REC.1400.095 در کمیته اخلاق دانشگاه آزاد اسلامی تهران شمال مورد تصویب قرار گرفته است.
نوع مطالعه: پژوهشي |
موضوع مقاله:
انتقال خون
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
