

جلد 20، شماره 2 - ( تابستان 1402 )
جلد 20 شماره 2 صفحات 177-155 |
برگشت به فهرست نسخه ها

![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Rajabi A, Ahmadinejad M. Acquired von Willebrand Syndrome. bloodj 2023; 20 (2) :155-177
URL: http://bloodjournal.ir/article-1-1478-fa.html
URL: http://bloodjournal.ir/article-1-1478-fa.html
رجبی علی، احمدینژاد مینو. سندروم فونویلبراند اکتسابی. فصلنامه پژوهشی خون. 1402; 20 (2) :155-177


دانشیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون
متن کامل [PDF 889 kb]
(928 دریافت)
| چکیده (HTML) (2131 مشاهده)
مقدمه
بیماری فونویلبراند(von Willebrand’s disease, vWD) نخستین بار در سال 1924 توسط محقق فنلاندی اریک آدولف فونویلبراند در یک دختر 5 ساله اهل فنلاند و سپس افراد زیادی از خانواده وی شناسایی و شرح داده شد. او مشخص کرد که این بیماری با هموفیلی تفاوت دارد به همین دلیل نام "هموفیلی کاذب ارثی" (hereditary pseudohemophilia) را بر آن گذاشت. این بیماری به افتخار او نامگذاری گردید(1). سپس نیم قرن پس از کشف بیماری، در سال 1971 عامل پلاسمایی اصلاحکننده بیماری توسط محقق آمریکایی تئودور زیمرمان (Theodore Zimmerman) شناسایی شد و به نام فاکتور فونویلبراند (von Willebrand’s factor, vWF) نامگذاری گردید(2).
vWF بزرگترین پروتئین پلاسمایی و یکی از گلیکوپروتئینهای فاز حاد است که توسط سلولهای اندوتلیال، مگاکاریوسیتها و بافت همبند زیر اندوتلیوم تولید میشود. هر مولکول vWF از چندین زیر واحد یکسان(مونومر) تشکیل شده است که در نهایت به صورت مولتیمرهایی حداکثر با 40 مونومر و اندازه 500 تا 20000 کیلودالتون در گردش خون دیده میشود. این اختلاف اندازه به دلیل تعداد متغیر مونومرهای تشکیلدهنده آن
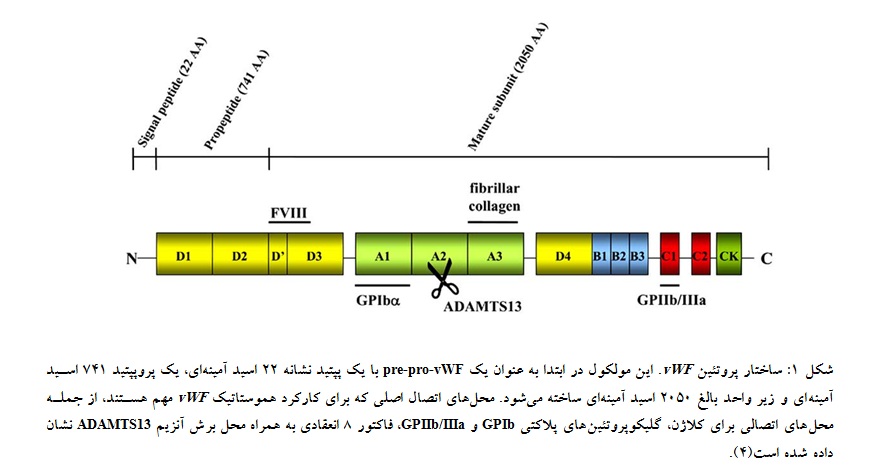
است. هر مونومر vWF دارای محل اتصال برای کلاژن و گلیکوپروتئینهای پلاکتی GPIb و GPIIb/IIIa است(شکل 1)(3).
vWF نقش محوری در هموستاز اولیه(با واسطه پلاکتها) و ثانویه(با واسطه فاکتورهای انعقادی) ایفا میکند. در هنگام آسیب عروقی و در محل ضایعه، برهمکنشهای متعدد جایگاههای تکراری اتصالی در مولتیمرهای vWF با پروتئینهای چسبنده زیراندوتلیوم (کلاژن) و نیز با گیرندههای سطح پلاکتی منجر به اتصال برگشتناپذیر پلاکتها با اندوتلیوم در معرض(exposed endothelium) میشود. با این کار، vWF به چسبندگی (adhesion) و تجمع(aggregation) پلاکتها کمک میکند. هم چنین با اتصال به فاکتور 8 انعقادی، آن را در گردش خون حمل کرده و به محل انعقاد خون میرساند. به علاوه، سبب پایدارسازی و محافظت فاکتور 8 و جلوگیری از تخریب آن میشود(6، 5). البته نقش فاکتور vWF محدود به پلاکتها نیست و نشان داده شده که در فرایندهای زیستی مختلفی همانند پیامدهی سلولی (cellular signaling)، رگزایی(angiogenesis) و التهاب نیز نقش دارد(7).
آنزیروتئولیتیک در تنظیم فیزیولوژیک اندازه vWF پلیمر نقش دارند. به طور مشخص، آنزیم متالوپروتئیناز ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13) مولتیمرهای بزرگ vWF را به محض ترشح به پلاسما به مولتیمرهای کوچکتر میشکند. محل شکست پروتئولیتیک vWF در ناحیه باند Tyr1605-Met1606 در دامین A2 قرار دارد(شکل 1)(9، 8). این شکست پروتئولیتیک برای تسهیل اتصال vWF به سلولها و پروتئینهای مختلف لازم است. پروتئازهای یاد شده در پاتوژنز اختلالات هموستازی و ترومبوزی ارثی و اکتسابی نقش مهمی ایفا میکنند. به عنوان نمونه عدم حضور مولتیمرهای بزرگ که در تیپ A2 بیماری و نیز در اختلالات میلوپرولیفراتیو به دلیل افزایش فعالیت ADAMTS13 دیده میشود، با تمایل به خونریزی همراه است و برعکس، وجود مولتیمرهای با اندازه فوقالعاده بزرگ که در TTP (Thrombotic Thrombocytopenic purpura) و HUS (Hemolytic Uremic Syndrome) به دلیل فعالیت ضعیف یا عدم فعالیت ADAMTS13 دیده میشود، با افزایش خطر ترومبوز همراه است(11-9). همچنین لازم به ذکر است که فعالیت چسبندگی vWF به اندوتلیوم وابسته به اندازه مولتیمرها است. هر چه مولتیمرها بزرگتر باشند، (High molecular weight multimers, HMWMs) فعالیت اتصالی آنها به پلاکتها و کلاژن بیشتر است. بنابراین از دست رفتن مولتیمرهای HMWM سبب از دست رفتن فعالیت هموستاتیک vWF میشود. به همین دلیل تنها مولتیمرهای بزرگتر فاکتور ویلبراند از نظر هموستاتیک فعال هستند(12، 10).

vWD: بیماری فونویلبراند، PT: زمان پروترومبین (Prothrombin Time)، PTT: زمان ترومبوپلاستین نسبی (Partial Thromboplastin Time)، PFA-100: سنجشگر کارکرد پلاکتی 100 (Platelet Function Analyser-100)، vWF:Ag: سطح آنتیژنی فاکتور فونویلبراند (von Willebrand’s Factor Antigen)، vWF:Rco: فعالیت کوفاکتوری ریستوستین فاکتور فونویلبراند (von Willebrand’s Factor Ristocetin Cofactor)، vWF:CB: فعالیت اتصال به کلاژن فاکتور فونویلبراند (von Willebrand’ Factor Collagen Binding)، RIPA: آگلوتیناسیون پلاکتی وابسته به ریستوستین (Ristocetin Induced Platelet Agglutination)، HMWMs: مولتیمرهای فاکتور فونویلبراند با وزن مولکولی بالا (High Molecular Weight Multimers)- برگرفته از (27، 24، 23، 18، 10، 6، 5).

شکل 3: سنجش مولتیمری در بیماران فونویلبراند ارثی و اکتسابی و مقایسه با نمونه پلاسمای طبیعی(normal plasma, NP). الف- مقایسه سنجش مولتیمری در AvWS به علت بیماریهای قلبی - عروقی با vWD نوع A2 و B2. ب- AvWS به علت MGUS. ج) AvWS به علت MPDs (28).
RCo/Ag و CB/Ag دیده میشود(30). در شکل 3 چند نمونه از سنجش مولتیمری به صورت شماتیک در بیماران vWD و AvWS با انتشار طبیعی مولتیمرهای vWF در نمونه پلاسمای طبیعی مقایسه شده است(شکل 3).
4-4 سنجش اتوآنتیبادیها:
بر خلاف هموفیلی اکتسابی، تعداد کمی از موارد AvWS با سازوکار اتوآنتیبادی بر ضد فعالیت vWF مرتبط با پلاکت مشخص میشوند. با این وجود، به نظر میرسد حضور آنتیبادیهای نوترالیزان با تمایل شدیدتری به خونریزی همراه باشد و به همین دلیل غربال آنتیبادی باید در همه موارد AvWS انجام شود(104، 99، 93، 57).
متداولترین روش مورد استفاده برای جستجوی فعالیت بازدارنده علیه vWF بر اساس مطالعات پلاسمای مخلوط (mixing studies) پلاسمای بیمار با پلاسمای معمولی و انکوباسیون در دمای 37 درجه سانتیگراد و به دنبال آن اندازهگیری vWF:RCo و vWF:CB اضافی (residual) است. با این حال، مهارکنندهها به ندرت شناسایی میشوند و آنتیبادیهایی که به vWF متصل شده و پاکسازی پلاسمایی آن را بدون خنثی کردن فعالیت vWF تسریع میکنند، با این روش قابل شناسایی نیستند. سنجشهای الایزا نیز به عنوان یک گزینه مطرح هستند اما هنوز به اندازه کافی استانداردسازی نشدهاند(105، 99، 64، 57).
اتوآنتیبادیهای آنتی-vWF در پاتوژنز برخی بیماران AvWS به ویژه مبتلایان به اختلالات لنفوپرولیفراتیو(LPDs) نقش دارند و شناسایی آنها یک ممیزهی تشخیصی(pathognomonic) برای سازوکار اکتسابی کمبود vWF است. همچنین به نظر میرسد که حضور این اتوآنتیبادیها با افزایش تمایل به خونریزی در بیماران همراه است. با این حال این آنتیبادیها تنها در 14 درصد موارد مشکوک به AvWS شناسایی شدهاند و ویژگیهای کارکردی متنوعی را نشان میدهند. همچنین به نظر میرسد حضور اتوAbها با خونریزی شدیدتر همراه است(58، 30).
بـر خلاف هموفیلی اکتسابی که مهارکنندههای فاکتور 8 عملاً همیشه عامل بیماری میباشند و با ارزیابیهای آزمایشگاهی استاندارد قابل شناسایی هستند، تنها در تعداد کمی از بیماران AvWS میتوان Abهای خنثیکننده یا نوترالیزان را با استفاده از ارزیابیهای خنثیکنندگی vWF:Ag، vWF:RCo، vWF:CB یا RIPA و مطالعههای پلاسمای مخلوط شناسایی کرد. با این حال، این ارزیابیها از نظر تکنیکی مشکل بوده و قادر به شناسایی آنتیبادیهای غیرنوترالیزان مرتبط نیستند. بنابراین، شناسایی مهارکنندهها در درصد کمی از بیماران AvWS ضرورتاً به معنای عدم حضور اتوآنتیبادیها نیست، بلکه ممکن است منعکسکننده حضور اتوAbهای غیرنوترالیزان باشد که پاکسازی vWF را از جریان خون سرعت میبخشند بدون اینکه روی سنجههای کارکردی قابل اندازهگیری اثر مهارکنندگی داشته باشند(107، 106، 57).
افزون بر اتوآنتیبادیها ، یافتن یک پروتئین مونوکلونال توسط الکتروفورز پروتئینهای سرمی نیز برای تشخیص AvWS محل بحث است. همچنین برخی مواقع مهارکنندهها با vWF کمپلکسهای اشباع تشکیل میدهند و بنابراین مانع شناسایی خود میشوند، مگر این که با کمک حرارت یا دیگر روشهای فیزیکی یا شیمیایی کمپلکسها تجزیه شود. همچنین یک روش الایزا معرفی شده که طیف وسیعتری از آنتیبادیهای متصلشونده به vWF را شناسایی میکند. آنتیبادیها غیرنوترالیزان متصلشونده به vWF در بیماران LPDs و نیز دیگر اختلالات زمینهای گزارش شده و میتوان آنها را توسط ELISA شناسایی کرد هر چند هنوز هیچ ارزیابی استانداردسازی شدهای برای شناسایی آنها در دسترس نیست. لازم به ذکر است که vWF مشتق از پلاسما حاوی آنتیژن گروه خونی ABO است و نباید به عنوان آنتیژن برای شناسایی اتوAbها در ELISA استفاده شود. به دلیل اینکه حضور ایزوآگلوتینینها میتواند سبب نتایج مثبت کاذب گردد. با این حال، میتوان برای رفع این نقیصه از vWF انسانی نوترکیب بیانشده در سلولهای حیوانی کشتشده به عنوان ریجنت استفاده کرد(105، 30، 28).
بایـد بـه خاطر داشت که تشخیص AvWS نیاز به تغییر در همه پارامترهای گفته شده در بالا ندارد. به عنوان مثال در یک مطالعه، هیلمن و همکاران AvWS را در بیماران تحت MCS ارزیابی کردند، و تشخیص بیماری را بر مبنای سنجش مولتیمری بنا نهادند که HMWMs غایب بودند و نیز دستکم یکی از نسبتهای کارکردی vWF کاهشیافته بود(108). البته ترجیح این است که تمامی سنجشهای مربوط به فاکتور فونویلبراند از جمله vWF:Ag، vWF:RCo، vWF:CB و سنجشهای مولتیمری در بیماران مشکوک به AvWS انجام شود.
لازم بـه ذکـر اسـت که نشانههای بالینی و آزمایشگاهی مرتبط با AvWS در اختلالات مختلف به طور معمول باعث ایجاد بیماری فونویلبراند نوع 2 میشود، هر چند در برخی بیماریها و حالات مختلف، بیمـاری نــوع 1 و یا 3 هم گزارش شده است(109).
همچنین باید به یاد داشت که اختلالات مرتبط با AvWS در جمعیت به نسبت معمول هستند. بنابراین انجام آزمونهای غربالگری جمعیتی ضروری نیست و حتی میتواند منجر به کسب نتایج مثبت کاذب فراوان و در نتیجه درمانهای غیر ضروری گردد. بنابراین انجام آزمونهای تشخیصی در بیماران بیعلامت، باید بر مبنای قضاوت بالینی پزشک هر بیمار باشد(28).
5- نشانههای بالینی و مدیریت درمان بیماری:
نشانههای بالینی در نوع ارثی و اکتسابی بیماری ویلبراند به طور عمده یکسان هستند. بنابراین انجام آزمونهای غربالگری در بیماران دارای اختلالات زمینهای مرتبط با AvWS قبل از جراحی ماژور و دیگر مداخلات با خطر بالای خونریزی پیشنهاد میشود(29).
نشانههای بالینی این بیماری همانند نوع ارثی، به طور عمده با خونریزی از مخاط(غشاهای موکوسی) مجاری معدهای رودهای(گوارشی) و ادراری تناسلی همراه است. به علاوه هماتومهای بافت نرم و خونریزیهای پس از عمل جراحی نیز معمول هستند(34). با این حال، شدت نشانههای بالینی در AvWS بستگی به شدت بیماری زمینه دارد و با درمان بیماری زمینهای، این بیماری نیز از بین میرود. بنابراین پیشآگهی و نیز مدیریت درمان بیماری وابسته به پاتوفیزیولوژی زمینه مرتبط با بیماری است. با این حال، با وجود انجام مطالعههای متعدد در رابطه با پاتولوژی بیماری، همچنان مدیریت درمان در AvWS همانند تشخیص بیماری، به عنوان یک چالش برای متخصصین خون باقیمانده است. هیچ دستورالعمل مبتنی بر شواهدی برای مدیریت بیماری وجود ندارد. درمان بیماریهای زمینهای بسته به بیماری به وسیله کورتیکواستروئیدها، شیمیدرمانی، سرکوبگرهای ایمنی، رادیوتراپی، جراحی و یا جایگزینی دریچه قلبی(vavle replacement) در بیماران نقص دریچههای قلبی میتوانند منجر به بهبودی در گروهی از بیماران شوند، اما این درمانها همیشه امکانپذیر و یا موفق نیستند. در مواردی که این درمانها به سرعت مؤثر نباشد، درمان علامتی با هدف اصلاح کمبود vWF به کار میرود تا از خونریزی غیرطبیعی جلوگیری و یا آنرا درمان کند. برخی از این گزینههای درمانی مانند دسموپرسین، کنسانترههای حاوی vWF، کنسانترههای فاکتور 8 نوترکیب(فاقد vWF)، فاکتور 7 نوترکیب فعال(rFVIIa)، IV-Ig و پلاسمافرز میتوانند در این مواقع مؤثر باشند. به خاطر ساز و کارهای مختلف AvWS ، اغلب بیش از یک رویکرد درمانی برای مدیریت خونریزیهای حاد و یا به عنوان پیشگیری در هنگام استفاده از روشهای تهاجمی در بیماران لازم است(95، 28، 25، 18).
بحث
ممکن است بر اساس شواهد تصور شود که بیماری فونویلبراند اکتسابی یک اختلال ناشایع است، اما بهتر است گفته شود که AvWS در واقع یک بیماری است که در بیشتر موارد یا کم تشخیص داده شده (underdiagnosed) و یا به اشتباه به عنوان نوع ارثی شناسایی میشود (misdiagnosed). هر چند تشخیص AvWS و تمایز بین نوع ارثی و اکتسابی بیماری بحثبرانگیز و دشوار است ولی تشخیص افتراقی AvWS از vWD ارثی به ویژه اشکال خفیفتر بیماری، با توجه به تفاوت در رویکرد درمانی مهم است. برای تشخیص نهایی AvWS ، نیاز به یافتههای مختلف بالینی و آزمایشگاهی است و باید در مطالعههای جامع، همه اطلاعات از جمله ارزیابی بالینی و تاریخچه موارد گزارش شده و نیز نتایج آزمایشگاهی را در نظر گرفت(57، 33، 30، 29).
آزمونهای آزمایشگاهی برای تشخیص AvWS و vWD ارثی یکسان هستند. بنابراین در حضور یک بیماری زمینهای مرتبط با AvWS، زمانی که یافتههای آزمایشگاهی و بالینی بیمار پیشنهاد دهنده بیماری فونویلبراند باشد، باید به ناچار تشخیص AvWS مد نظر قرار گیرد. همچنین گرفتن شرح حال از بیمار برای تشخیص AvWS مهم است.
باید دقت شود که اخذ تاریخچه با جزییات و معاینه جسمی کافی باشد تا بیشتر اختلالات مرتبط تحت پوشش قرار گیرند. معاینات به ویژه در مورد بیماریهای قلبی - عروقی مانند تنگی دریچه آئورت اهمیت زیادی دارند. به همین ترتیب بیشتر اختلالات هماتولوژیک، بدخیم و خودایمن باید به طور بالینی مشخص و غربال شوند. به علاوه الکتروفورز پروتئین سرمی و شمارش خون کامل (CBC) میتواند به ترتیب به غربال و شناسایی گاموپاتیهای مونوکلونال و دیگر اختلالات هماتولوژیک کمک کنند(110، 28، 24).
نتیجهگیری
با توجه به گستردگی بیماریهای مرتبط با AvWS، لازم است هم متخصصین بالینی به ویژه هماتولوژیستها و متخصصین قلب و هم متخصصین آزمایشگاه توجه ویژهای به این بیماری داشته باشند تا موارد AvWS به درستی شناسایی و تشخیص داده شوند. بنابراین برای همه بیماران دارای یافتههای آزمایشگاهی مرسوم، اختلالات فاکتور ویلبراند(vWF) به همراه تاریخچه منفی خونریزی پیشین در بیمار و یا سابقه خانوادگی وی، غربال برای اختلالات مرتبط با AvWS توصیه میشود. بنابراین تظاهر دیرهنگام حوادث خونریزی در یک زمینه با تاریخچه منفی خانوادگی باید شک را به سمت AvWS ببرد.
متن کامل: (2440 مشاهده)
سندروم فونویلبراند اکتسابی
علی رجبی1، مینو احمدینژاد2
چکیده
سابقه و هدف
بیماری فونویلبراند شایعترین اختلال ارثی خونریزی دهنده در دنیا است اما نوع اکتسابی بیماری که در زمینه برخی بیماریها دیده میشود، فراوانی کمتری دارد. بیماریهای زمینهای و استفاده از وسایل کمکی قلبی- عروقی، اختلالات لنفوپرولیفراتیو و میلوپرولیفراتیو از شایعترین بیماریهای همراه با نوع اکتسابی هستند. باوجود شیوع کم، نوع اکتسابی یک اختلال خونریزیدهنده مهم از نظر بالینی میباشد.
مواد و روشها
با توجه به اهمیت بالینی بیماری، در این نوشتار مروری با رجوع به پایگاههای اطلاعاتی PubMed، Medline، Scopus و Google Scholar و استفاده از کلید واژههای مربوطه، 110 مقاله مرتبط با نوع اکتسابی فون ویلبراند از زمان شناسایی بیماری تا زمان حاضر، در زمینههای تشخیص، یافتههای بالینی و آزمایشگاهی بیماری بررسی و مرور شد.
یافتهها
یافتههای بالینی و آزمایشگاهی در بیماری فونویبلبراند و نوع اکتسابی آن، اغلب شبیه به هم هستند و همین موضوع تشخیص بیماری را مشکل میکند. با این حال، انجام آزمونهای غربالگری در بیماران همراه با اختلالات زمینهای مرتبط با نوع اکتسابی قبل از مداخلات بالینی مانند جراحی که با خطر بالای خونریزی همراه هستند، تا حدود زیادی تشخیص بیماری را راحتتر میسازد. همچنین یک جنبه کلیدی تمایز نوع اکتسابی و بیماری فونویلبراند، رخداد این بیماری بدون وجود تاریخچه پیشین خونریزی در بیمار یا دیگر اعضای خانواده وی میباشد.
نتیجه گیری
بر اساس گزارشهای منتشر شده، نوع اکتسابی به عنوان یک اختلال با شیوع کم در نظر گرفته شده ولی به نظر میرسد که شیوع واقعی آن نسبت به آن چیزی که قبلاً تصور میشد، به دلیل آگاهی و شناخت بیشتر متخصصین به ویژه هماتولوژیستها و متخصصین قلب، دستکم در گروههای مشخصی از بیماران در حال افزایش است.
کلمات کلیدی: فاکتور فونویلبراند، اختلالات خونریزیدهنده، بیماری فونویلبراند
تاریخ دریافت: 21/09/1401
تاریخ پذیرش : 07/12/1401
1- PhD هماتولوژی و بانک خون ـ مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
2- مؤلف مسئول: متخصص آسیبشناسی بالینی ـ دانشیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران ـ صندوق پستی: 1157-14665
علی رجبی1، مینو احمدینژاد2
چکیده
سابقه و هدف
بیماری فونویلبراند شایعترین اختلال ارثی خونریزی دهنده در دنیا است اما نوع اکتسابی بیماری که در زمینه برخی بیماریها دیده میشود، فراوانی کمتری دارد. بیماریهای زمینهای و استفاده از وسایل کمکی قلبی- عروقی، اختلالات لنفوپرولیفراتیو و میلوپرولیفراتیو از شایعترین بیماریهای همراه با نوع اکتسابی هستند. باوجود شیوع کم، نوع اکتسابی یک اختلال خونریزیدهنده مهم از نظر بالینی میباشد.
مواد و روشها
با توجه به اهمیت بالینی بیماری، در این نوشتار مروری با رجوع به پایگاههای اطلاعاتی PubMed، Medline، Scopus و Google Scholar و استفاده از کلید واژههای مربوطه، 110 مقاله مرتبط با نوع اکتسابی فون ویلبراند از زمان شناسایی بیماری تا زمان حاضر، در زمینههای تشخیص، یافتههای بالینی و آزمایشگاهی بیماری بررسی و مرور شد.
یافتهها
یافتههای بالینی و آزمایشگاهی در بیماری فونویبلبراند و نوع اکتسابی آن، اغلب شبیه به هم هستند و همین موضوع تشخیص بیماری را مشکل میکند. با این حال، انجام آزمونهای غربالگری در بیماران همراه با اختلالات زمینهای مرتبط با نوع اکتسابی قبل از مداخلات بالینی مانند جراحی که با خطر بالای خونریزی همراه هستند، تا حدود زیادی تشخیص بیماری را راحتتر میسازد. همچنین یک جنبه کلیدی تمایز نوع اکتسابی و بیماری فونویلبراند، رخداد این بیماری بدون وجود تاریخچه پیشین خونریزی در بیمار یا دیگر اعضای خانواده وی میباشد.
نتیجه گیری
بر اساس گزارشهای منتشر شده، نوع اکتسابی به عنوان یک اختلال با شیوع کم در نظر گرفته شده ولی به نظر میرسد که شیوع واقعی آن نسبت به آن چیزی که قبلاً تصور میشد، به دلیل آگاهی و شناخت بیشتر متخصصین به ویژه هماتولوژیستها و متخصصین قلب، دستکم در گروههای مشخصی از بیماران در حال افزایش است.
کلمات کلیدی: فاکتور فونویلبراند، اختلالات خونریزیدهنده، بیماری فونویلبراند
تاریخ دریافت: 21/09/1401
تاریخ پذیرش : 07/12/1401
1- PhD هماتولوژی و بانک خون ـ مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
2- مؤلف مسئول: متخصص آسیبشناسی بالینی ـ دانشیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران ـ صندوق پستی: 1157-14665
مقدمه
بیماری فونویلبراند(von Willebrand’s disease, vWD) نخستین بار در سال 1924 توسط محقق فنلاندی اریک آدولف فونویلبراند در یک دختر 5 ساله اهل فنلاند و سپس افراد زیادی از خانواده وی شناسایی و شرح داده شد. او مشخص کرد که این بیماری با هموفیلی تفاوت دارد به همین دلیل نام "هموفیلی کاذب ارثی" (hereditary pseudohemophilia) را بر آن گذاشت. این بیماری به افتخار او نامگذاری گردید(1). سپس نیم قرن پس از کشف بیماری، در سال 1971 عامل پلاسمایی اصلاحکننده بیماری توسط محقق آمریکایی تئودور زیمرمان (Theodore Zimmerman) شناسایی شد و به نام فاکتور فونویلبراند (von Willebrand’s factor, vWF) نامگذاری گردید(2).
vWF بزرگترین پروتئین پلاسمایی و یکی از گلیکوپروتئینهای فاز حاد است که توسط سلولهای اندوتلیال، مگاکاریوسیتها و بافت همبند زیر اندوتلیوم تولید میشود. هر مولکول vWF از چندین زیر واحد یکسان(مونومر) تشکیل شده است که در نهایت به صورت مولتیمرهایی حداکثر با 40 مونومر و اندازه 500 تا 20000 کیلودالتون در گردش خون دیده میشود. این اختلاف اندازه به دلیل تعداد متغیر مونومرهای تشکیلدهنده آن
است. هر مونومر vWF دارای محل اتصال برای کلاژن و گلیکوپروتئینهای پلاکتی GPIb و GPIIb/IIIa است(شکل 1)(3).
vWF نقش محوری در هموستاز اولیه(با واسطه پلاکتها) و ثانویه(با واسطه فاکتورهای انعقادی) ایفا میکند. در هنگام آسیب عروقی و در محل ضایعه، برهمکنشهای متعدد جایگاههای تکراری اتصالی در مولتیمرهای vWF با پروتئینهای چسبنده زیراندوتلیوم (کلاژن) و نیز با گیرندههای سطح پلاکتی منجر به اتصال برگشتناپذیر پلاکتها با اندوتلیوم در معرض(exposed endothelium) میشود. با این کار، vWF به چسبندگی (adhesion) و تجمع(aggregation) پلاکتها کمک میکند. هم چنین با اتصال به فاکتور 8 انعقادی، آن را در گردش خون حمل کرده و به محل انعقاد خون میرساند. به علاوه، سبب پایدارسازی و محافظت فاکتور 8 و جلوگیری از تخریب آن میشود(6، 5). البته نقش فاکتور vWF محدود به پلاکتها نیست و نشان داده شده که در فرایندهای زیستی مختلفی همانند پیامدهی سلولی (cellular signaling)، رگزایی(angiogenesis) و التهاب نیز نقش دارد(7).
آنزیروتئولیتیک در تنظیم فیزیولوژیک اندازه vWF پلیمر نقش دارند. به طور مشخص، آنزیم متالوپروتئیناز ADAMTS13 (a disintegrin-like and metalloprotease with thrombospondin type 1 motif 13) مولتیمرهای بزرگ vWF را به محض ترشح به پلاسما به مولتیمرهای کوچکتر میشکند. محل شکست پروتئولیتیک vWF در ناحیه باند Tyr1605-Met1606 در دامین A2 قرار دارد(شکل 1)(9، 8). این شکست پروتئولیتیک برای تسهیل اتصال vWF به سلولها و پروتئینهای مختلف لازم است. پروتئازهای یاد شده در پاتوژنز اختلالات هموستازی و ترومبوزی ارثی و اکتسابی نقش مهمی ایفا میکنند. به عنوان نمونه عدم حضور مولتیمرهای بزرگ که در تیپ A2 بیماری و نیز در اختلالات میلوپرولیفراتیو به دلیل افزایش فعالیت ADAMTS13 دیده میشود، با تمایل به خونریزی همراه است و برعکس، وجود مولتیمرهای با اندازه فوقالعاده بزرگ که در TTP (Thrombotic Thrombocytopenic purpura) و HUS (Hemolytic Uremic Syndrome) به دلیل فعالیت ضعیف یا عدم فعالیت ADAMTS13 دیده میشود، با افزایش خطر ترومبوز همراه است(11-9). همچنین لازم به ذکر است که فعالیت چسبندگی vWF به اندوتلیوم وابسته به اندازه مولتیمرها است. هر چه مولتیمرها بزرگتر باشند، (High molecular weight multimers, HMWMs) فعالیت اتصالی آنها به پلاکتها و کلاژن بیشتر است. بنابراین از دست رفتن مولتیمرهای HMWM سبب از دست رفتن فعالیت هموستاتیک vWF میشود. به همین دلیل تنها مولتیمرهای بزرگتر فاکتور ویلبراند از نظر هموستاتیک فعال هستند(12، 10).
vWF در سلولهای اندوتلیال و مگاکاریوسیتها به عنوان یک پیشپروتئین(pre-pro-vWF) به طول 2813 نوکلئوتید به شکل مونومر ساخته میشود که دارای یک پپتید نشانه(signal peptide) با 22 اسیدآمینه، یک پروپپتید (vWF propeptide, vWFpp)(که قبلاً به نام آنتیژن II فونویلبراند خوانده میشد) با 741 اسیدآمینه و vWF بالغ با 2050 اسیدآمینه میباشد(شکل 1). پروپپتید و زیر واحد بالغ، مولکول pro-vWF متشکل از 2791 اسیدآمینه و 4 نوع دامنه تکراری(repeated domains) را تشکیل میدهند. پس از حذف پپتید نشانه از pre-pro-vWF، pro-vWF به شبکه آندوپلاسمی خشن منتقل میشود و در آن جا زیر واحدهای pro-vWF با تشکیل پیوندهای دیسولفیدی بین دامنههای CK غنی از سیستئین در انتهای کربوکسیل(C) مولکول به صورت دُمبهدُم(tail-to-tail) تشکیل دایمر میدهند و سپس به دستگاه گلژی منتقل میشوند. در گلژی، دایمرهای متصلشده دُمبهدُم با تشکیل پیوندهای دیسولفیدی سربهسر(head-to-head) بین دامنههای D3 غنی از سیستئین در انتهای آمینی(N) مولکول، مولتیمریزه شده و دستخوش تغییرات گستردهی پس از ترجمه (post-translational modification) میشوند(شکل 1). vWF در نهایت به عنوان یک پروتئین بالغ در اجسام وایبلپالادِ سلولهای اندوتلیال(Weibel-Palade bodies)، مگاکاریوسیتها و پلاکتها(گرانولهای آلفا) بستهبندی، ذخیره و ترشح میگردد. سلولهای اندوتلیال به طور اولیه vWF را ترشح میکنند، اما پلاکتها تنها در هنگام تحریک شدن آن را رها مینمایند(15-13، 4، 3).
مانکوزو و همکارانش درسال 1989 گزارش کردند که ژن vWF دارای 178 کیلوباز طول و 52 اگزون میباشد. این ژن روی بازوی کوتاه کروموزوم 12 قرار دارد. طول اگزونها از 40 تا 1379 جفتباز و اینترونها از 97 جفتباز تا تقریباً 9/19 کیلوباز متغیر هستند. پپتید نشانه و پروپپتید فاکتور فونویلبراند توسط 17 اگزون در تقریباً 80 کیلو باز از DNA ژن vWF رمزگذاری میشوند در حالی که زیرواحد بالغ فاکتور فونویلبراند و ناحیه غیررمزکننده '3 توسط 35 اگزون در حدود 100 کیلوباز باقیمانده رمزگذاری میشود(16).
بیماری فونویلبراند:
vWD شایعترین اختلال خونریزیدهنده ارثی با شیوع تقریبی از 6/0 تا 3/1 درصد(بر اساس مطالعات جمعیتی) و با میانگین 1% در جمعیت عمومی میباشد، اما نوع علامتدار بیماری شیوع 01/0 درصد در جوامع انسانی دارد. این بیماری به علت اختلال کمّی یا کیفی در vWF ایجاد میشود و به صورت اتوزومی به ارث میرسد(17، 5).
نشانههای بالینی بیماری به طور عمده دربرگیرنده خونریزیهای پوستیـ مخاطی خفیف تا متوسط و نیز خونریزیهای غیر عادی پس از جراحی، تروما و یا حتی بریدگیهای کوچک است. با این حال، بسته به نوع و شدت بیماری ممکن است از یک اکیموز(کبودی) ساده خود به خودی(easy bruising) بر روی دست و پای بیمار تا خون دماغ، خونریزیهای لثه و در خانمها افزایش خونریزیهای قاعدگی از خفیف تا شدید(منوراژی) و یا افزایش خونریزی در حین و پس از زایمان را در برگیرد. خونریزیهای شدیدتر و یا عمیقتر از جمله خونریزیهای معدهای رودهای(دستگاه گوارش)، هماتومها و هماتروز، به جز در نوع شدید بیماری غیر معمول است. vWD هر دو جنس مذکر و مؤنث را به یک اندازه مبتلا میکند اما در خانمها ممکن است به دلیل خونریزیهای قاعدگی و بعد از زایمان شیوع بیشتری داشته باشد(18، 17).
به طور کلی تشخیص اولیه vWD بر اساس سابقه شخصی یا خانوادگی از کاهش vWF و شواهد آزمایشگاهی از اختلالات کمی یا کیفی vWF، فاکتور VIII و یا هر دو داده میشود. بیماران مبتلا، دارای سطوح کاهشیافته کارکردی vWF میباشند. به طور معمول vWD ممکن است در هنگام یک چالش هموستاتیک و یا به طور اتفاقی در طی آزمایش چکاپ آشکار شود و ممکن است با افزایش سن و مواجهه بیشتر با حوادث، نشانههای خونریزی بیشتر آشکار شود. دستورالعملهای جدید توصیه کردهاند که سطح vWF IU/dL 30 به عنوان مرز جداساز (cutoff) برای شناسایی اختلال vWD در نظر گرفته شود و افراد دارای سطح vWF بالای 30 و کمتر از 50 نیز با دارا بودن نشانههای خونریزی جزو این گروه محسوب میشوند. این تغییر در دستورالعملها به طور برجستهای تعداد افراد مبتلا به هر کدام از انواع بیماری را تغییر داده است(24-19).
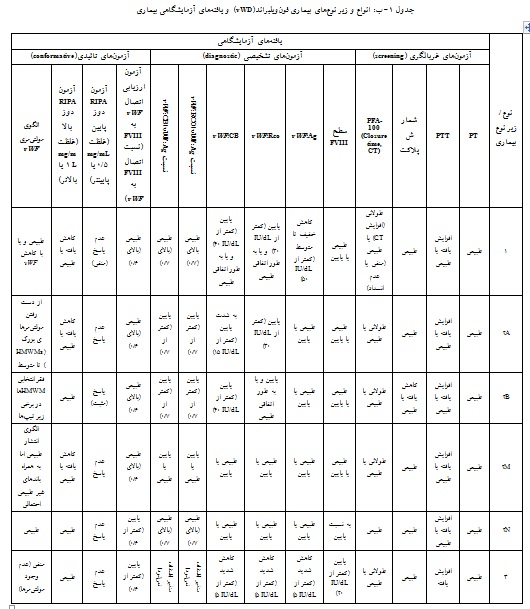
بیماری فونویلبراند به دو صورت ارثی و اکتسابی وجود دارد. شکل ارثی بیماری(vWD) میتواند هم به صورت اتوزوم غالب و هم مغلوب به ارث برسد و به چهار نوع کلی 1، 2، 3 و پلاکتی دستهبندی میشود. انواع مختلف بیماری را میتوان بر اساس ویژگیهای فنوتیپی از هم افتراق داد(26، 25). در جدول انوع زیرگروههای ارثی vWD با مشخصههای بالینی و آزمایشگاهی مربوطه ذکر شده است(جدول 1).
نوع 1 بیماری یک اختلال کمّی است که با کمبود خفیف vWF (سطح آنتیژنی vWF:Ag کمتر از IU/dL 30)، خونریزیهای پوستی مخاطی خفیف تا متوسط و گاهی خونریزیهای شدیدتر پس از جراحی تظاهر میکند و حدود 80%-60% (میانگین حدود 70%) موارد بیماری را در برمیگیرد. این اختلال میتواند به طور اتفاقی طی آزمایش روتین و یا اعمال جراحی خفیف(همانند کشیدن دندان، جراحی لثه و یا برداشتن لوزه) شناسایی گردد.
نوع 2 بیماری با اختلال کیفی و متوسط vWF و خونریزی پوستی مخاطی خفیف تا متوسط همراه بوده و مسئول حدود 20 تا 30 درصد کل بیماران vWD است. این نوع خود به چهار زیرنوع(subtype) A2، B2، M2 و N2 دستهبندی میشود.
نوع A2 بیماری به طور معمول با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و نبود مولتیمرهای متوسط و بزرگ vWF (HMWMs) به دلیل تجمع(assembly) ناقص مولتیمرها و افزایش حساسیت به شکست توسط ADAMTS13 مشخص میشود.
نوع B2 به طور معمول با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و ترومبوسیتوپنی که در بعضی شرایط خاص بدتر میشود تظاهر میکند. در این نوع، کمبود HMWMs ثانویه به نقایص کارکردی vWF به دلیل اتصال خود به خودی vWF به GPIb پلاکتها دیده میشود. این تمایل افزایش یافته vWF برای اتصال به پلاکتها سبب پاکسازی کمپلکسهای vWF-پلاکت توسط سیستم رتیکولواندوتلیال میگردد. یک نقص مشابه افزایش کارکرد(gain of function) در GPIb پلاکتی نیز باعث vWD نوع پلاکتی میشود که با افزایش اتصال خودبهخود پلاکتها به vWF در جریان خون و در نهایت پاکسازی مولکولهای vWF میشود. نوع M2 بیماری نیز با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و نیز نقص اتصال کلاژن (collagen binding) و توزیع و تجمع طبیعی مولتیمرهای vWF بروز میکند.
در نــوع N2 بیماری، نقص در اتصال vWF به فاکتور 8
انعقادی سبب تخریب سریع این فاکتور و در نتیجه سطوح
پایین فاکتور 8 میشود که میتواند سبب تظاهر بیشتر خونریزی به ویژه پس از جراحی شده و نشانههایی شبیه هموفیلی خفیف تا متوسط A را تقلید کند.
نوع 3 بیماری شدیدترین نوع بیماری بوده که در جوامع انسانی بسیار نادر است(کمتر از 3 در هر میلیون نفر) و کمتر از 10 درصد موارد vWD را تشکیل میدهد. نوع 3 با خونریزیهای شدید پوستی - مخاطی و اسکلتی-ماهیچهای(musculoskeletal) و کمبود شدید (vWF:Ag کمتر از IU/dL 5) یا فقدان سطح آنتیژنی vWF بروز میکند (جدول 1 الف و ب)(27، 24، 23، 18، 10، 6، 5).
بیماری فونویلبراند اکتسابی:
بر خلاف نوع ارثی بیماری(vWD) که شایعترین اختلال
ارثی خونریزیدهنده است، نوع اکتسابی بیماری (Acquired von Willebrand’s Syndrome, AvWS) نادر میباشد، هرچند به نظر میرسد در بیشتر موارد، بیماری کم تشخیص داده شده(underdiagnosed) و یا به اشتباه به عنوان نوع ارثی (vWD) تشخیص داده میشود (misdiagnosed). در نتیجه شیوع آن غالباً کمتر از واقعیت تخمین زده میشود(underestimated). این موضوع احتمالاً به خاطر گستره وسیع ویژگیهای بالینی و آزمایشگاهی بیماری است. با این حال بیماری فونویلبراند اکتسابی از نظر بالینی یک اختلال خونریزیدهنده مهم است. مطالعههای مختلف گزارش شده بیانگر این موضوع هستند که احتمال ارجاع بیماران مبتلا به AvWS به متخصصین به ویژه هماتولوژیستها و متخصصین قلب و در نتیجه شیوع بیماری نسبت به گذشته در حال افزایش است(28، 5).
مانکوزو و همکارانش درسال 1989 گزارش کردند که ژن vWF دارای 178 کیلوباز طول و 52 اگزون میباشد. این ژن روی بازوی کوتاه کروموزوم 12 قرار دارد. طول اگزونها از 40 تا 1379 جفتباز و اینترونها از 97 جفتباز تا تقریباً 9/19 کیلوباز متغیر هستند. پپتید نشانه و پروپپتید فاکتور فونویلبراند توسط 17 اگزون در تقریباً 80 کیلو باز از DNA ژن vWF رمزگذاری میشوند در حالی که زیرواحد بالغ فاکتور فونویلبراند و ناحیه غیررمزکننده '3 توسط 35 اگزون در حدود 100 کیلوباز باقیمانده رمزگذاری میشود(16).
بیماری فونویلبراند:
vWD شایعترین اختلال خونریزیدهنده ارثی با شیوع تقریبی از 6/0 تا 3/1 درصد(بر اساس مطالعات جمعیتی) و با میانگین 1% در جمعیت عمومی میباشد، اما نوع علامتدار بیماری شیوع 01/0 درصد در جوامع انسانی دارد. این بیماری به علت اختلال کمّی یا کیفی در vWF ایجاد میشود و به صورت اتوزومی به ارث میرسد(17، 5).
نشانههای بالینی بیماری به طور عمده دربرگیرنده خونریزیهای پوستیـ مخاطی خفیف تا متوسط و نیز خونریزیهای غیر عادی پس از جراحی، تروما و یا حتی بریدگیهای کوچک است. با این حال، بسته به نوع و شدت بیماری ممکن است از یک اکیموز(کبودی) ساده خود به خودی(easy bruising) بر روی دست و پای بیمار تا خون دماغ، خونریزیهای لثه و در خانمها افزایش خونریزیهای قاعدگی از خفیف تا شدید(منوراژی) و یا افزایش خونریزی در حین و پس از زایمان را در برگیرد. خونریزیهای شدیدتر و یا عمیقتر از جمله خونریزیهای معدهای رودهای(دستگاه گوارش)، هماتومها و هماتروز، به جز در نوع شدید بیماری غیر معمول است. vWD هر دو جنس مذکر و مؤنث را به یک اندازه مبتلا میکند اما در خانمها ممکن است به دلیل خونریزیهای قاعدگی و بعد از زایمان شیوع بیشتری داشته باشد(18، 17).
به طور کلی تشخیص اولیه vWD بر اساس سابقه شخصی یا خانوادگی از کاهش vWF و شواهد آزمایشگاهی از اختلالات کمی یا کیفی vWF، فاکتور VIII و یا هر دو داده میشود. بیماران مبتلا، دارای سطوح کاهشیافته کارکردی vWF میباشند. به طور معمول vWD ممکن است در هنگام یک چالش هموستاتیک و یا به طور اتفاقی در طی آزمایش چکاپ آشکار شود و ممکن است با افزایش سن و مواجهه بیشتر با حوادث، نشانههای خونریزی بیشتر آشکار شود. دستورالعملهای جدید توصیه کردهاند که سطح vWF IU/dL 30 به عنوان مرز جداساز (cutoff) برای شناسایی اختلال vWD در نظر گرفته شود و افراد دارای سطح vWF بالای 30 و کمتر از 50 نیز با دارا بودن نشانههای خونریزی جزو این گروه محسوب میشوند. این تغییر در دستورالعملها به طور برجستهای تعداد افراد مبتلا به هر کدام از انواع بیماری را تغییر داده است(24-19).
بیماری فونویلبراند به دو صورت ارثی و اکتسابی وجود دارد. شکل ارثی بیماری(vWD) میتواند هم به صورت اتوزوم غالب و هم مغلوب به ارث برسد و به چهار نوع کلی 1، 2، 3 و پلاکتی دستهبندی میشود. انواع مختلف بیماری را میتوان بر اساس ویژگیهای فنوتیپی از هم افتراق داد(26، 25). در جدول انوع زیرگروههای ارثی vWD با مشخصههای بالینی و آزمایشگاهی مربوطه ذکر شده است(جدول 1).
نوع 1 بیماری یک اختلال کمّی است که با کمبود خفیف vWF (سطح آنتیژنی vWF:Ag کمتر از IU/dL 30)، خونریزیهای پوستی مخاطی خفیف تا متوسط و گاهی خونریزیهای شدیدتر پس از جراحی تظاهر میکند و حدود 80%-60% (میانگین حدود 70%) موارد بیماری را در برمیگیرد. این اختلال میتواند به طور اتفاقی طی آزمایش روتین و یا اعمال جراحی خفیف(همانند کشیدن دندان، جراحی لثه و یا برداشتن لوزه) شناسایی گردد.
نوع 2 بیماری با اختلال کیفی و متوسط vWF و خونریزی پوستی مخاطی خفیف تا متوسط همراه بوده و مسئول حدود 20 تا 30 درصد کل بیماران vWD است. این نوع خود به چهار زیرنوع(subtype) A2، B2، M2 و N2 دستهبندی میشود.
نوع A2 بیماری به طور معمول با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و نبود مولتیمرهای متوسط و بزرگ vWF (HMWMs) به دلیل تجمع(assembly) ناقص مولتیمرها و افزایش حساسیت به شکست توسط ADAMTS13 مشخص میشود.
نوع B2 به طور معمول با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و ترومبوسیتوپنی که در بعضی شرایط خاص بدتر میشود تظاهر میکند. در این نوع، کمبود HMWMs ثانویه به نقایص کارکردی vWF به دلیل اتصال خود به خودی vWF به GPIb پلاکتها دیده میشود. این تمایل افزایش یافته vWF برای اتصال به پلاکتها سبب پاکسازی کمپلکسهای vWF-پلاکت توسط سیستم رتیکولواندوتلیال میگردد. یک نقص مشابه افزایش کارکرد(gain of function) در GPIb پلاکتی نیز باعث vWD نوع پلاکتی میشود که با افزایش اتصال خودبهخود پلاکتها به vWF در جریان خون و در نهایت پاکسازی مولکولهای vWF میشود. نوع M2 بیماری نیز با خونریزی پوستی- مخاطی، نقص کارکرد پلاکتی وابسته به vWF و نیز نقص اتصال کلاژن (collagen binding) و توزیع و تجمع طبیعی مولتیمرهای vWF بروز میکند.
در نــوع N2 بیماری، نقص در اتصال vWF به فاکتور 8
انعقادی سبب تخریب سریع این فاکتور و در نتیجه سطوح
پایین فاکتور 8 میشود که میتواند سبب تظاهر بیشتر خونریزی به ویژه پس از جراحی شده و نشانههایی شبیه هموفیلی خفیف تا متوسط A را تقلید کند.
نوع 3 بیماری شدیدترین نوع بیماری بوده که در جوامع انسانی بسیار نادر است(کمتر از 3 در هر میلیون نفر) و کمتر از 10 درصد موارد vWD را تشکیل میدهد. نوع 3 با خونریزیهای شدید پوستی - مخاطی و اسکلتی-ماهیچهای(musculoskeletal) و کمبود شدید (vWF:Ag کمتر از IU/dL 5) یا فقدان سطح آنتیژنی vWF بروز میکند (جدول 1 الف و ب)(27، 24، 23، 18، 10، 6، 5).
بیماری فونویلبراند اکتسابی:
بر خلاف نوع ارثی بیماری(vWD) که شایعترین اختلال
ارثی خونریزیدهنده است، نوع اکتسابی بیماری (Acquired von Willebrand’s Syndrome, AvWS) نادر میباشد، هرچند به نظر میرسد در بیشتر موارد، بیماری کم تشخیص داده شده(underdiagnosed) و یا به اشتباه به عنوان نوع ارثی (vWD) تشخیص داده میشود (misdiagnosed). در نتیجه شیوع آن غالباً کمتر از واقعیت تخمین زده میشود(underestimated). این موضوع احتمالاً به خاطر گستره وسیع ویژگیهای بالینی و آزمایشگاهی بیماری است. با این حال بیماری فونویلبراند اکتسابی از نظر بالینی یک اختلال خونریزیدهنده مهم است. مطالعههای مختلف گزارش شده بیانگر این موضوع هستند که احتمال ارجاع بیماران مبتلا به AvWS به متخصصین به ویژه هماتولوژیستها و متخصصین قلب و در نتیجه شیوع بیماری نسبت به گذشته در حال افزایش است(28، 5).
AvWS به علت نقایص ساختاری یا کارکردی vWF و ثانویه به اختلالات مختلف ایجاد میشود که بیماریهای قلبی- عروقی و اختلالات لنفوپرولیفراتیو و میلوپرولیفراتیو ازشایعترین عوامل ایجاد بیماری محسوب میشوند. اختلالات دیگر مانند بیماریهای ایمونولوژیک، دیگر بدخیمیها، بیماریهای خودایمن، اختلالات مختلف دیگر، و نیز برخی مداخلات درمانی مانند تجویز عوامل ضد تشنج و یا استفاده از دستگاههای پشتیبانی مکانیکی گردش خون (mechanical circulatory support, MCS) به ویژه در بیماران قلبی- عروقی از عوامل نادر ایجاد AvWS در بیماران محسوب میشوند(جدول 1)(30، 29). با توجه به مقدمه ذکر شده و اهمیت تشخیص این بیماری و به دلیل مغفول ماندن این بیماری در بین متخصصین کشور و نیز جای خالی مطالعه جامعی در این زمینه، مقاله حاضر با هدف مرور جامعی بر بیماری یا سندروم فونویلبراند اکتسابی نگارش شد تا متخصصین بالینی و آزمایشگاهی با دید بهتری به این بیماری نگاه کرده و موارد بالینی واقعی مرتبط با بیماری با سهولت بیشتری شناسایی و درمان گردند.
مواد و روشها
در این نوشتار، به مروری بر بیماری فونویلبراند اکتسابی، تاریخچه و یافتههای بالینی و آزمایشگاهی و روش تشخیص بیماری میپردازیم. در تهیه منابع نوشتار، با رجوع به پایگاههای اطلاعاتی PubMed، Medline، Scopus و Google Scholar، با توجه به کلید واژههای مربوط به بیماری فونویلبراند بر اساس MeSH، و هم چنین استفاده از واژگان ترکیبی و تکی von Willebrand’s disease، von Willebrand’s factor،deficincy von Willebrand’s factor، acquired von Willebrand’s disease، acquired von Willebrand’s syndrome، acquired bleeding disorders و acquired blood coagulation disorders، با استفاده از عملگرهای[AND] و [OR] جستجو انجام گردید. سپس با استفاده از همین کلید واژهها و همراهی واژه Iran ، وضعیت بیماری در ایران جستجو شد. با توجه به نادر بودن بیماری و کشف اولین مورد در سال 1968 میلادی، جستجو بدون محدودیت زمانی تا سال 2022 انجام شد. سپس مقالات جستجو شده با توجه به عنوان مقالات و بررسی چکیدهها پالایش گردید. در مرحله تدوین مطالب، ابتدا سرفصلهای مورد نظر برای مطالعه انتخاب و با توجه به آن مقالات نهایی انتخاب گردید. در نهایت تعداد 110 مقاله در این مطالعه مورد استفاده و استناد قرار گرفت.
یافتهها
1- تاریخچه و اپیدمیولوژی بیماری:
1-1 تاریخچه:
اولین مورد AvWS در سال 1968 توسط جوزف سیمون در یک کودک مبتلا به بیماری لوپوس اریتماتوز سیستمیک(Systemic Lupus Erythematosus, SLE) گزارش شد. تشخیص AvWS برای بیمار در سن 13 سالگی داده شده بود و یک سال قبل از تشخیص، در سن 12 سالگی شروع به خون دماغ(اپیستاکسی) مکرر کرده بود. سپس نشانههای خونریزی ناشی از ساییدگی کام سخت(abrasion of the hard palate) و پس از آن کبودیهای خودبهخود(easy bruising) در بیمار واضحتر از قبل رخ میداد. در اواسط سن 13 سالگی با کشیدن دندان، خونریزی طولانی به مدت سه روز و سپس بستری در بیمارستان با تزریق خون کامل و بستن حفره دندان(dental socket) همراه شد. خونریزی تا روز دهم بستری قطع نشد. مطالعههای آزمایشگاهی در آن زمان نشان داد که آزمونهای زمان لختگی(clotting time, CT)، زمان پروترومبین(prothrombin ime, PT)، تعداد پلاکتها، جمع شدن لخته(clot retraction) و مصرف پروترومبین (prothrombin consumption) طبیعی، و زمان ترومبوپلاستین نسبی(partial thromboplastin time, PTT) به طور قابل توجهی طولانی شده است.
تعداد گلبولهای سفید در طول بستری از 2900 تا 6100 متغیر بود. بیمار یک ماه بعد در کلینیک هماتولوژی اطفال ویزیت شد و تشخیص بیماری فونویلبراند بر اساس شرح حال بالا، زمان خونریزی(bleeding time, BT) طولانی، PTT غیرطبیعی و سطح فاکتور VIII انعقادی 17% داده شد. والدین و شش خواهر و برادر او سالم بودند و هیچ سابقه خونریزی غیرعادی یا بیماریهای خودایمنی نداشتند. بیمار نیز تا 14 سالگی وضعیت بالینی مناسبی داشت، تا زمانی که اولین واکسن آبله را به او تزریق کردند. یک هفته پس از واکسیناسیون، با درد در شانه چپ و بزرگ و حساس شدن گرههای لنفاوی زیر بغل سمت چپ و گرفتگی در محل واکسیناسیون(بازوی چپ) ویزیت شد. دو ماه بعد نشانههای قرمزی و پوستهپوسته شدن صورت، پیشانی، گوشها و بینی، نشانههای اندامی از جمله گلومرولونفریت کلیوی و در نهایت مثبت شدن آزمون لوپوس(از جمله سلولهای LE)، آنتیبادیهای ضد هسته سرم و ضد انعقادهای گردشی دیده شد.
در نهایـت تشخیـص نهایـی SLE با همراهی بیماری فونویلبراند (AvWS) برای بیمار داده شده و درمان با کورتیکواستروئیدها از جمله پردنیزون و تزریق خون آغاز شد. بیمار رفتهرفته در طی چند سال درمان و تا سن 19 سالگی به طور نسبی بهبود یافت و به تدریج نشانههای بالینی و آزمونهای آزمایشگاهی (هماتولوژیک و انعقادی) او از جمله بیماری فونویلبراند و سلولهای LE، طبیعی و ناپدید شدند و بیمار تا این سن و زمان گزارش بیماری (سال 1968) بدون علامت باقی ماند (31).
2-1 اپیدمیولوژی و شیوع بیماری:
پس از گزارش نخست سیمون، اینگرام و همکاران در سال 1971 چهار مورد دیگر AvWS را گزارش کردند که یک مورد مبتلا به SLE بود و با درمان استروئیدی به سرعت بهبود یافت. در سه بیمار دیگر هرچند بررسیهای متعددی انجام شد، اما هیچ علامتی از بیماری زمینهای یافت نشده و با استروئیدها درمان نشدند(32).
در مجموع از زمان شناسایی اولیه بیماری در سال 1968 تا سال 1999، تعداد 266 مورد AvWS در 116 مقاله گزارش شد که عمده اینها گزارش تکموردی بودند. سپس در یک بررسی گذشتهنگر توسط جامعه بینالمللی ترومبوز و هموستاز(International Society on Thrombosis and Haemostasis, ISTH) در فاصله سالهای 1998 و 1999 که در سال 2000 منتشرشد، 186 مورد دیگر گزارش شد (29). این گزارش جامعترین مطالعه بینالمللی تاکنون در مورد AvWS محسوب میشود.
در سال 2002، باده و همکاران نتایج مطالعهای را گزارش کردند که در یک دوره بیش از 2 سال، نمونه پلاسمای 5014 بیمار مبتلا به اختلالات خونریزیدهنده از بیش از 80 بیمارستان و آزمایشگاه در سراسر آلمان به آزمایشگاهی در هامبورگ ارسال شده و ارزیابیهای بعدی به منظور تشخیص vWD بر روی این نمونهها انجام شد. در نهایت از میان این بیماران، 187 مورد جدید AvWS گزارش شد (33).
به دلیل نبود دادههای آیندهنگر بزرگ و یا مطالعههای اپیدمیولوژیک درباره AvWS تا به امروز، شیوع واقعی بیماری در جمعیت عمومی هنوز مشخص نشده و فقط یک تخمین تقریبی در دسترس ما است(34). در کل، موارد گزارش شده AvWS تاکنون کمتر از 1000 مورد در سراسر دنیا بوده است. همچنین از آن جا که موارد خفیفتر بیماری فقط در طی چالشهای هموستاتیک تشخیص داده میشوند و نیز به دلیل عدم آگاهی پزشکان متخصص از این بیماری، این اختلال به درستی تشخیص داده نشده و شیوع آن کم تخمین زده میشود. تقریباً چیزی تا حدود 5/4 درصد بیمارانی که در آنها تشخیص بیماری فونویلبراند داده شده، ممکن است مبتلا به نوع اکتسابی بیماری باشند. با توجه به شیوع vWD در جوامع انسانی (حدود 1%)، تخمین زده میشود که در حال حاضر شیوع AvWS دامنه ای از 04/0 تا 13/0 درصد در جمعیت عمومی داشته باشد(36، 35، 28). با این حال، شیوع آن در گروههای خاصی بیشتر است. به عنوان مثال، این بیماری تا 20% از موارد بدخیمیها و تا 100% برخی از اعمال قلبی مانند دریچههای فلزی قلب(metallic cardiac valves) و اکسیژنرسانی غشایی برونپیکری (خارج از بدن) extracorporeal membrane oxygenation, ECMO)) گزارش شده است(40-37).
3-1 دامنه سنی ابتلا:
بر اساس انتشار سنی اختلالات زمینه، AvWS میتواند در هر سنی از نوزادی تا کهنسالی(دامنه سنی 2 تا 96 سال) رخ دهد ولی تظاهر اولیه بیماری به طور معمول در بالغین و بیشتر در سنین دهه 7 و 8 زندگی(با میانگین سنی ابتلای 62 سال) و بدون سابقه شخصی یا خانوادگی از تمایل به خونریزی بروز میکند. به طور معمول بیماری در کودکان نادرتر از بزرگسالان است. با این حال در برخی مطالعهها گزارش شده که AvWS در سنین کودکی به ویژه در کودکان مبتلا به نقایص قلبی ارثی میتواند یافتهی شایعــی
باشد(41، 37، 30، 29).
2- ساز و کار بیماریزایی:
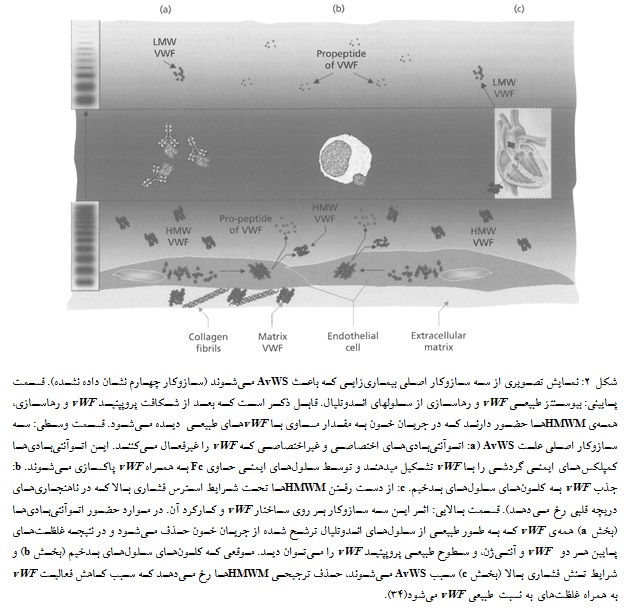
اختلالات زمینهای که سبب ایجاد ناهنجاریهای vWF و در نهایت بیماری AvWS میشوند، با چندین ساز و کار مختلف این کار را انجام میدهند(شکل 2):
1-2 پاکسازی (clearance) به واسطه کمپلکس اتوآنتیبادی با vWF :
اتوآنتیبادیهای ضد vWF دربرگیرنده مهارکنندههای جایگاههای کارکردی یا ساختاری مولکول همراه با تداخل در کارکرد آن از جمله اتوآنتیبادیهای مداخلهکننده با پلاکتها و متصلشونده به کلاژن(collagen binding) و نیز اتوآنتیبادیهای غیرمهارکننده میباشند. این آنتیبادیها با vWF کمپلکس ایمنی تشکیل میدهند و منجر به پاکسازی سریع این مولکول از گردش خون میشوند. این ساز و کار بیشتر در AvWS مرتبط با گاموپاتیهای مونوکلونال و بیماریهای خودایمن دیده میشود.
2-2 جذب vWF به سطوح پلاکتی و نیز کلونهای سلولهای تغییرشکلیافته یا بدخیم مثل سلولهای میلوما:
در نتیجه جذب vWF به سطوح پلاکتی و سلولهای بدخیم، احتباس HMWMها رخ میدهد. این ساز و کار در بیماران با اختلالات هماتولوژیک و نیز در ترومبوسیتوز واکنشی اثبات شده است.
3-2 افزایش تخریب HMWMs :
به دلیل افزایش تنش برشی(shear stress) و متعاقب آن پروتئولیز vWF رخ میدهد. AvWS در نتیجه این ساز و کار در شرایط هموراژیک غیرطبیعی مانند ناهنجاریهای شکلی قلبی- عروقی مثل تنگی دریچه آئورت و دستگاههای کمکی بطن چپ با جریان مداوم(Left Ventricular Assist Device, LVAD) توصیف شده است. شکست پروتئولیتیک vWF همچنین در بیماران با پانکراتیت، سیروز کبدی، لوسمی و نیز تجویز یکسری داروهای خاص نیز توصیف شده است.
4-2 کاهش سنتز vWF :
به نظر میرسد که در برخی اختلالات مانند هیپوتیروئیدی، AvWS در اثر کاهش سنتز vWF ایجاد میشود(47-42، 30، 28).
3- علل ایجاد بیماری:
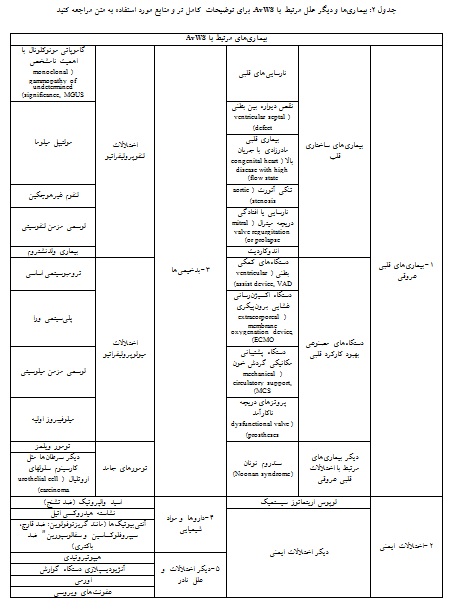
AvWS به طور معمول با یک اختلال زمینهای مرتبط است که طیف زیادی از بیماریهای مختلف را درگیر میکنند. با این حال تعداد کمی از بیماریهای مختلف، درصد زیادی از اختلالات مرتبط با AvWS را در بر میگیرند(جدول 2)(46).
مواد و روشها
در این نوشتار، به مروری بر بیماری فونویلبراند اکتسابی، تاریخچه و یافتههای بالینی و آزمایشگاهی و روش تشخیص بیماری میپردازیم. در تهیه منابع نوشتار، با رجوع به پایگاههای اطلاعاتی PubMed، Medline، Scopus و Google Scholar، با توجه به کلید واژههای مربوط به بیماری فونویلبراند بر اساس MeSH، و هم چنین استفاده از واژگان ترکیبی و تکی von Willebrand’s disease، von Willebrand’s factor،deficincy von Willebrand’s factor، acquired von Willebrand’s disease، acquired von Willebrand’s syndrome، acquired bleeding disorders و acquired blood coagulation disorders، با استفاده از عملگرهای[AND] و [OR] جستجو انجام گردید. سپس با استفاده از همین کلید واژهها و همراهی واژه Iran ، وضعیت بیماری در ایران جستجو شد. با توجه به نادر بودن بیماری و کشف اولین مورد در سال 1968 میلادی، جستجو بدون محدودیت زمانی تا سال 2022 انجام شد. سپس مقالات جستجو شده با توجه به عنوان مقالات و بررسی چکیدهها پالایش گردید. در مرحله تدوین مطالب، ابتدا سرفصلهای مورد نظر برای مطالعه انتخاب و با توجه به آن مقالات نهایی انتخاب گردید. در نهایت تعداد 110 مقاله در این مطالعه مورد استفاده و استناد قرار گرفت.
یافتهها
1- تاریخچه و اپیدمیولوژی بیماری:
1-1 تاریخچه:
اولین مورد AvWS در سال 1968 توسط جوزف سیمون در یک کودک مبتلا به بیماری لوپوس اریتماتوز سیستمیک(Systemic Lupus Erythematosus, SLE) گزارش شد. تشخیص AvWS برای بیمار در سن 13 سالگی داده شده بود و یک سال قبل از تشخیص، در سن 12 سالگی شروع به خون دماغ(اپیستاکسی) مکرر کرده بود. سپس نشانههای خونریزی ناشی از ساییدگی کام سخت(abrasion of the hard palate) و پس از آن کبودیهای خودبهخود(easy bruising) در بیمار واضحتر از قبل رخ میداد. در اواسط سن 13 سالگی با کشیدن دندان، خونریزی طولانی به مدت سه روز و سپس بستری در بیمارستان با تزریق خون کامل و بستن حفره دندان(dental socket) همراه شد. خونریزی تا روز دهم بستری قطع نشد. مطالعههای آزمایشگاهی در آن زمان نشان داد که آزمونهای زمان لختگی(clotting time, CT)، زمان پروترومبین(prothrombin ime, PT)، تعداد پلاکتها، جمع شدن لخته(clot retraction) و مصرف پروترومبین (prothrombin consumption) طبیعی، و زمان ترومبوپلاستین نسبی(partial thromboplastin time, PTT) به طور قابل توجهی طولانی شده است.
تعداد گلبولهای سفید در طول بستری از 2900 تا 6100 متغیر بود. بیمار یک ماه بعد در کلینیک هماتولوژی اطفال ویزیت شد و تشخیص بیماری فونویلبراند بر اساس شرح حال بالا، زمان خونریزی(bleeding time, BT) طولانی، PTT غیرطبیعی و سطح فاکتور VIII انعقادی 17% داده شد. والدین و شش خواهر و برادر او سالم بودند و هیچ سابقه خونریزی غیرعادی یا بیماریهای خودایمنی نداشتند. بیمار نیز تا 14 سالگی وضعیت بالینی مناسبی داشت، تا زمانی که اولین واکسن آبله را به او تزریق کردند. یک هفته پس از واکسیناسیون، با درد در شانه چپ و بزرگ و حساس شدن گرههای لنفاوی زیر بغل سمت چپ و گرفتگی در محل واکسیناسیون(بازوی چپ) ویزیت شد. دو ماه بعد نشانههای قرمزی و پوستهپوسته شدن صورت، پیشانی، گوشها و بینی، نشانههای اندامی از جمله گلومرولونفریت کلیوی و در نهایت مثبت شدن آزمون لوپوس(از جمله سلولهای LE)، آنتیبادیهای ضد هسته سرم و ضد انعقادهای گردشی دیده شد.
در نهایـت تشخیـص نهایـی SLE با همراهی بیماری فونویلبراند (AvWS) برای بیمار داده شده و درمان با کورتیکواستروئیدها از جمله پردنیزون و تزریق خون آغاز شد. بیمار رفتهرفته در طی چند سال درمان و تا سن 19 سالگی به طور نسبی بهبود یافت و به تدریج نشانههای بالینی و آزمونهای آزمایشگاهی (هماتولوژیک و انعقادی) او از جمله بیماری فونویلبراند و سلولهای LE، طبیعی و ناپدید شدند و بیمار تا این سن و زمان گزارش بیماری (سال 1968) بدون علامت باقی ماند (31).
2-1 اپیدمیولوژی و شیوع بیماری:
پس از گزارش نخست سیمون، اینگرام و همکاران در سال 1971 چهار مورد دیگر AvWS را گزارش کردند که یک مورد مبتلا به SLE بود و با درمان استروئیدی به سرعت بهبود یافت. در سه بیمار دیگر هرچند بررسیهای متعددی انجام شد، اما هیچ علامتی از بیماری زمینهای یافت نشده و با استروئیدها درمان نشدند(32).
در مجموع از زمان شناسایی اولیه بیماری در سال 1968 تا سال 1999، تعداد 266 مورد AvWS در 116 مقاله گزارش شد که عمده اینها گزارش تکموردی بودند. سپس در یک بررسی گذشتهنگر توسط جامعه بینالمللی ترومبوز و هموستاز(International Society on Thrombosis and Haemostasis, ISTH) در فاصله سالهای 1998 و 1999 که در سال 2000 منتشرشد، 186 مورد دیگر گزارش شد (29). این گزارش جامعترین مطالعه بینالمللی تاکنون در مورد AvWS محسوب میشود.
در سال 2002، باده و همکاران نتایج مطالعهای را گزارش کردند که در یک دوره بیش از 2 سال، نمونه پلاسمای 5014 بیمار مبتلا به اختلالات خونریزیدهنده از بیش از 80 بیمارستان و آزمایشگاه در سراسر آلمان به آزمایشگاهی در هامبورگ ارسال شده و ارزیابیهای بعدی به منظور تشخیص vWD بر روی این نمونهها انجام شد. در نهایت از میان این بیماران، 187 مورد جدید AvWS گزارش شد (33).
به دلیل نبود دادههای آیندهنگر بزرگ و یا مطالعههای اپیدمیولوژیک درباره AvWS تا به امروز، شیوع واقعی بیماری در جمعیت عمومی هنوز مشخص نشده و فقط یک تخمین تقریبی در دسترس ما است(34). در کل، موارد گزارش شده AvWS تاکنون کمتر از 1000 مورد در سراسر دنیا بوده است. همچنین از آن جا که موارد خفیفتر بیماری فقط در طی چالشهای هموستاتیک تشخیص داده میشوند و نیز به دلیل عدم آگاهی پزشکان متخصص از این بیماری، این اختلال به درستی تشخیص داده نشده و شیوع آن کم تخمین زده میشود. تقریباً چیزی تا حدود 5/4 درصد بیمارانی که در آنها تشخیص بیماری فونویلبراند داده شده، ممکن است مبتلا به نوع اکتسابی بیماری باشند. با توجه به شیوع vWD در جوامع انسانی (حدود 1%)، تخمین زده میشود که در حال حاضر شیوع AvWS دامنه ای از 04/0 تا 13/0 درصد در جمعیت عمومی داشته باشد(36، 35، 28). با این حال، شیوع آن در گروههای خاصی بیشتر است. به عنوان مثال، این بیماری تا 20% از موارد بدخیمیها و تا 100% برخی از اعمال قلبی مانند دریچههای فلزی قلب(metallic cardiac valves) و اکسیژنرسانی غشایی برونپیکری (خارج از بدن) extracorporeal membrane oxygenation, ECMO)) گزارش شده است(40-37).
3-1 دامنه سنی ابتلا:
بر اساس انتشار سنی اختلالات زمینه، AvWS میتواند در هر سنی از نوزادی تا کهنسالی(دامنه سنی 2 تا 96 سال) رخ دهد ولی تظاهر اولیه بیماری به طور معمول در بالغین و بیشتر در سنین دهه 7 و 8 زندگی(با میانگین سنی ابتلای 62 سال) و بدون سابقه شخصی یا خانوادگی از تمایل به خونریزی بروز میکند. به طور معمول بیماری در کودکان نادرتر از بزرگسالان است. با این حال در برخی مطالعهها گزارش شده که AvWS در سنین کودکی به ویژه در کودکان مبتلا به نقایص قلبی ارثی میتواند یافتهی شایعــی
باشد(41، 37، 30، 29).
2- ساز و کار بیماریزایی:
اختلالات زمینهای که سبب ایجاد ناهنجاریهای vWF و در نهایت بیماری AvWS میشوند، با چندین ساز و کار مختلف این کار را انجام میدهند(شکل 2):
1-2 پاکسازی (clearance) به واسطه کمپلکس اتوآنتیبادی با vWF :
اتوآنتیبادیهای ضد vWF دربرگیرنده مهارکنندههای جایگاههای کارکردی یا ساختاری مولکول همراه با تداخل در کارکرد آن از جمله اتوآنتیبادیهای مداخلهکننده با پلاکتها و متصلشونده به کلاژن(collagen binding) و نیز اتوآنتیبادیهای غیرمهارکننده میباشند. این آنتیبادیها با vWF کمپلکس ایمنی تشکیل میدهند و منجر به پاکسازی سریع این مولکول از گردش خون میشوند. این ساز و کار بیشتر در AvWS مرتبط با گاموپاتیهای مونوکلونال و بیماریهای خودایمن دیده میشود.
2-2 جذب vWF به سطوح پلاکتی و نیز کلونهای سلولهای تغییرشکلیافته یا بدخیم مثل سلولهای میلوما:
در نتیجه جذب vWF به سطوح پلاکتی و سلولهای بدخیم، احتباس HMWMها رخ میدهد. این ساز و کار در بیماران با اختلالات هماتولوژیک و نیز در ترومبوسیتوز واکنشی اثبات شده است.
3-2 افزایش تخریب HMWMs :
به دلیل افزایش تنش برشی(shear stress) و متعاقب آن پروتئولیز vWF رخ میدهد. AvWS در نتیجه این ساز و کار در شرایط هموراژیک غیرطبیعی مانند ناهنجاریهای شکلی قلبی- عروقی مثل تنگی دریچه آئورت و دستگاههای کمکی بطن چپ با جریان مداوم(Left Ventricular Assist Device, LVAD) توصیف شده است. شکست پروتئولیتیک vWF همچنین در بیماران با پانکراتیت، سیروز کبدی، لوسمی و نیز تجویز یکسری داروهای خاص نیز توصیف شده است.
4-2 کاهش سنتز vWF :
به نظر میرسد که در برخی اختلالات مانند هیپوتیروئیدی، AvWS در اثر کاهش سنتز vWF ایجاد میشود(47-42، 30، 28).
3- علل ایجاد بیماری:
AvWS به طور معمول با یک اختلال زمینهای مرتبط است که طیف زیادی از بیماریهای مختلف را درگیر میکنند. با این حال تعداد کمی از بیماریهای مختلف، درصد زیادی از اختلالات مرتبط با AvWS را در بر میگیرند(جدول 2)(46).
بر اساس مطالعه 186 فرد مبتلا به AvWS در گزارش ثبتی ISTH، اختلالات لنفوپرولیفراتیو با 48% و بیماریهای قلبی- عروقی با 21% موارد، شایعترین عوامل ایجاد AvWS گزارش شدند. اختلالات میلوپرولیفراتیو با 15%، دیگر نئوپلاسمها با 5% و بیماریهای ایمونولوژیک (خودایمنی) با 2% شیوع، از دیگر بیماریهای شایع زمینهای بودند (29). در گزارشهای مختلف بسته به جمعیت مورد مطالعه ممکن است میزان شیوع AvWS در بیماریهای زمینهای مختلف، آمار متفاوتی ذکر شود. به عنوان مثال علیرغم گزارش ISTH که اختلالات لنفوپرولیفراتیو را شایعترین عامل AvWS معرفی کرده بود، اکثر منابع اختلالات قلبی عروقی را شایعترین بیماریهای زمینهای در میان اختلالات متعدد مرتبط با AvWS گزارش نمودند(37)(جدول 2). تیده و همکاران نیز نشان دادند که در بین تمام بیماران مبتلا به AvWS، این بیماری در 46% موارد با اختلالات قلبی عروقی در ارتباط بود(30).
1-3 اختلالات قلبی- عروقی:
در مطالعههای اخیر، AvWS و بیماریهای قلبی - عروقی روند ارتباطی فزایندهای را نشان میدهند(میزان
1-3 اختلالات قلبی- عروقی:
در مطالعههای اخیر، AvWS و بیماریهای قلبی - عروقی روند ارتباطی فزایندهای را نشان میدهند(میزان
شیوع 46%-40%). این موضوع بیانگر آگاهی روزافزون متخصصین و جراحان قلب از علل و زمینههای ایجاد AvWS و در نتیجه ارجاع بیماران بیشتری برای تشخیص میباشد(33، 30).
علاوه بر بیماریهای ساختاری قلب، با استفاده روزافزون از دستگاههای MCS برای نارساییهای قلبی، AvWS نیز به طور مکرر در بیماران استفاده کننده از این ابزارها دیده میشود. به همین علت، مشکلات خونریزی مرتبط با AvWS علت اصلی ناخوشی و مرگ و میر (morbidity and mortality) در این بیماران ذکر شده است(37). افزون بر این، تشخیص AvWS در بیماران مبتلا به تنگی دریچه آئورت شدید تا 79% و LVAD تا 100% بیماران به همراه شیوع بالای خونریزی گزارش شده است(50-48). با این حال، تکرر خونریزی در این بیماران با سطح vWF:Ag یا vWF:RCo مرتبط نیست. لازم به ذکر است که با افزایش تعداد بیماران نارسایی قلبی که به مدت طولانی در انتظار پیوند نگهداری میشوند، LVAD توجهات را در خصوص یک اختلال زمینهای مهم مرتبط با AvWS به خود جلب کرده است(51، 28). با این حال هر چند که گزارشهایی در این زمینه به طور عمده روی تنگی دریچه آئورت و LVAD تمرکز کردهاند، اما باید یادآوری شود که دامنه گستردهای از نقایص قلبی ارثی و اکتسابی در ارتبـاط بـا AvWS گـزارش شـده اســت، مانند پروتزهای دریچه ناکارآمد (dysfunctional valve prostheses)، اندوکاردیت و نقایص دیوارهای بطنی قلبی(septal defects)(49، 30، 29).
بیماریهای قلبی- عروقی مختلف مانند تنگی آئورت (aortic stenosis)، کاردیومیوپاتی انسدادی هیپرتروفیک (hypertrophic obstructive cardiomyopathy) و برخی از دیگر بیماریهای ساختاری مادرزادی قلبی و نیز MCS،
علاوه بر بیماریهای ساختاری قلب، با استفاده روزافزون از دستگاههای MCS برای نارساییهای قلبی، AvWS نیز به طور مکرر در بیماران استفاده کننده از این ابزارها دیده میشود. به همین علت، مشکلات خونریزی مرتبط با AvWS علت اصلی ناخوشی و مرگ و میر (morbidity and mortality) در این بیماران ذکر شده است(37). افزون بر این، تشخیص AvWS در بیماران مبتلا به تنگی دریچه آئورت شدید تا 79% و LVAD تا 100% بیماران به همراه شیوع بالای خونریزی گزارش شده است(50-48). با این حال، تکرر خونریزی در این بیماران با سطح vWF:Ag یا vWF:RCo مرتبط نیست. لازم به ذکر است که با افزایش تعداد بیماران نارسایی قلبی که به مدت طولانی در انتظار پیوند نگهداری میشوند، LVAD توجهات را در خصوص یک اختلال زمینهای مهم مرتبط با AvWS به خود جلب کرده است(51، 28). با این حال هر چند که گزارشهایی در این زمینه به طور عمده روی تنگی دریچه آئورت و LVAD تمرکز کردهاند، اما باید یادآوری شود که دامنه گستردهای از نقایص قلبی ارثی و اکتسابی در ارتبـاط بـا AvWS گـزارش شـده اســت، مانند پروتزهای دریچه ناکارآمد (dysfunctional valve prostheses)، اندوکاردیت و نقایص دیوارهای بطنی قلبی(septal defects)(49، 30، 29).
بیماریهای قلبی- عروقی مختلف مانند تنگی آئورت (aortic stenosis)، کاردیومیوپاتی انسدادی هیپرتروفیک (hypertrophic obstructive cardiomyopathy) و برخی از دیگر بیماریهای ساختاری مادرزادی قلبی و نیز MCS،
با افزایش تنش برشی در جریان خون سبب افزایش شکافت مولتیمرهای vWF و در نتیجه از دست دادن HMWMs میشوند.
در این بیماران به طور معمول، vWF:Ag، vWF:RCo و vWF:CB در سطح طبیعی هستند و یا حتی افزایش دارند. نسبتهای vWF:RCo/Ag و vWF:CB/Ag اغلب اما نه همیشه، کاهشیافته هستند. در برخی بیماران، کاهش یا فقدان HMWMs تنها ناهنجاری آزمایشگاهی نشاندهنده AvWS است. بنابراین فقدان HMWMs به تنهایی میتواند یک فاکتور خطر خونریزی در بیماران قلبی عروقی محسوب شود(54-52، 50، 48، 39). علاوه بر بیماریهای زمینهای قلبی عروقی، AvWS در دیگر بیماریهایی که با اختلالات قلبی همراه هستند مانند سندروم نونان(Noonan syndrome) نیز گزارش شده است (56، 55).
2-3 بدخیمیها:
1-2-3 اختلالات لنفوپرولیفراتیو:
اختلالات مختلف لنفوپرولیفراتیو(LPDs ، Lymphoproliferative ( مسئول بخش قابل توجهی از موارد AvWS هم در گزارشهای مختلف منتشر شده(30%) و هم در گزارش ثبتی ISTH (48%) است(57، 29). در یک مطالعه آیندهنگر توسط موهری و همکاران، 8 مورد از 113 بیمار(7%) مبتلا به LPDs، با AvWS همراه بودند که در50% بیماران خونریزی بالینی اثبات شد(58). در یک مطالعه دیگر توسط تیده و همکاران، 11 مورد از 35 بیمار AvWS مورد مطالعه (31%)، در نتیجه گاموپتی مونوکلونال از جمله میلوم متعدد (multiple myeloma) و ماکروگلوبولینمی ولدنشتروم (Waldenström’s disease) بودند که قبل از تشخیص AvWS با خونریزی تظاهر کرده بودند(30). البته باید یادآوری شود که خونریزی در نتیجه AvWS در LPDs به تنهایی رخ نمیدهد، مثلاً در بیماران مبتلا به بیماری ولدنشتروم یا IgM-MGUS (monoclonal gammopathy of undetermined significance)، خونریزی میتواند حاصل از افزایش ویسکوزیته خون باشد(62-59، 30). دیگر اختلالات LPDs مانند لوسمی مزمن لنفوسیتی (chronic lymphocytic leukemia, CLL) و لنفوم غیر هوچکین(non-Hodgkin lymphoma, NHL) و دیگر لنفومها مثل لنفوم لنفوپلاسماسیتیک (Lymphoplasmacytic Lymphoma) و آمیلوئیدوز نیز با شیوع کمتری به عنوان عامل AvWS معرفی شدهاند(66-63).
2-2-3 نئوپلاسمهای میلوپرولیفراتیو:
AvWS مرتبـط بـا نئـوپـلاسـمهـای میلوپرولیفراتیـو (myeloproliferative neoplasia, MPNs) بـه طـور اولیه در نتیجه جذب سطحی vWF به سلولهای خونی تغییر شکل یافته به ویژه پلاکتها حاصل میشود(67، 57). این موضوع در ترومبوسیتمی اساسی (essential thrombocythemia, ET) و پلیسیتمی ورا (polycythemia vera, PV)، همچنین در لوسمی مزمن میلوئیدی (chronic myelocytic leukemia, CML)، میلوفیبروز اولیه (primary myelofibrosis, PM) و برخی مواقع در لوسمیهای حاد گزارش شده است(72-68). در میان MPNs، شیوع AvWS در ET شایعتر از بقیه گزارش شده است (57).
در مطالعه موهری و همکاران، AvWS در 14 مورد از 125 بیمار مبتلا به اختلالات MPN (11%) گزارش گردید (58).
لازم به ذکر است که فاکتورهای خطر خونریزی در MPN به خوبی توصیف نشده است، اما بیشتر مطالعهها گزارش کردهاند که شمارش پلاکتی بسیار بالا(بیشتر از /μL103×1500) یک فاکتور خطر با اهمیت برای ایجاد خونریزی است(73). در دو مطالعه گزارش شده توسط ون ژندرن و همکاران در سال 1994، از بین 100 بیمار با ترومبوسیتمی هموراژیک، میانگین ± انحراف معیار شمارش پلاکتی nL/2016±1070 بود. بر همین اساس، یک ارتباط معکوس بین شمارش پلاکتی و نسبت vWF:RCo/Ag یا vWF:CB/Ag در بیماران MPN گزارش گردید(28). با این حال، در مطالعه آیندهنگر موهری و همکاران، شمارش پلاکت میانه(median) بیماران مبتلا به MPN و AvWS تنها /nL638 (با دامنه /nL120-1305) بود (58). بنابراین این موضوع به نظر محتمل میرسد که عوامل خطر دیگر مثل نقایص کارکردی پلاکتها نیز در خونریزی مشارکت دارند. با این حال، ارتباط بین شمارش پلاکتی، AvWS ، نقایص کارکردی پلاکتها و خطر خونریزی مستلزم مطالعه بیشتر است. بنابراین پیشنهاد میشود که بیماران خونریزیدهنده با MPN به منظور هر دو اختلال کارکردی پلاکتی و AvWS غربال شوند(73، 39).
3-3 دیگر اختلالات مرتبط با AvWS :
اختـلالات دیگـر مرتبـط با AvWS بسیار متعدد هستند ولی درصد کمی از علل ایجاد کننده بیماری را دربر میگیرند. هر چند نخستین گزارش AvWS در SLE گزارش شده است، اما اختلالات ایمنی و خود ایمن مثل- SLE ، آرتریت روماتوئید، اختلالات بافت همبند مانند بیماری بافت همبند مختلط(mixed connective tissue disease) و نشانگان اهلرز-دنلوس (Ehlers–Danlos syndromes)، سندروم آنتیفسفولیپید (antiphospholipid antibody syndrome)، حضور ضد انعقادهای شبه لوپوسی(lupus-like anticoagulants)، سندروم شوگرن(Sjogren's syndrome)، اسکلروز منتشر(systemic sclerosis) یا اسکلرودرما (scleroderma)، اختلال استخوانزایی(Osteogenesis Imperfecta) و اختلال دومفصلی خوشخیم(Benign Joint Hypermobility Syndrome) با شیوع کمتری به عنوان عامل ایجاد AvWS گزارش شدهاند(83-74، 49).
به غیر از اختلالات LPDs و MPNs ، بدخیمیهای دیگر مانند تومور ویلمز، ماستوسیتوز منتشر(systemic mastocytosis)، اختلالات متابولیکی و ذخیرهای مانند هیپوتیروئیدی، اورمی، بیماری گوشه و آنژیودیسپلازی دستگاه گوارش با شیوع کمتری به عنوان عامل AvWS معرفی شدهاند(90-84، 33).
به علاوه برخی از داروها از قبیل اسید والپروئیک (ضد تشنج)، نشاسته هیدروکسی اتیل(به عنوان انبساط دهنده پلاسما در موارد شوک هیپوولمیک)، برخی آنتیبیوتیکها مانند گریزئوفولوین(ضد قارچ)، تتراسایکلین، سیپروفلوکساسین و سفالوسپورین(ضد باکتری)، عوامل ترومبوتیک و استفاده از آفتکشهــا نیز به عنوان علت نادر AvWS گزارش شدهاند(93-91).
4- تشخیص بیماری:
دستیابی تشخیصی به AvWS به خاطر تظاهر بالینی متغیر و آزمونهای مختلف مورد نیاز برای اثبات یا رد تشخیص آن چالشبرانگیز و مشکل است و اساساً هیچ آزمون واحدی برای اثبات یا رد تشخیص بیماری در دسترس نیست(93، 57، 28). نشانههای بالینی خونریزی در بیماران AvWS به طور عمده همانند نوع ارثی از جمله خونریزیهای پوستی- مخاطی میباشد ولی مشخصترین یافته بالینی این است که بیماران به طور عمده تاریخچه خونریزیدهنده در خود یا خانواده خود ندارند. دیگر یافتههای بالینی بستگی به نشانههای بیماری زمینهای دارد (93). در غیاب سابقه خانوادگی و یا فردی خونریزی، آزمونهای تشخیصی برای AvWS همان آزمونهای مورد استفاده برای تشخیص vWD ارثی هستند(94).
باید به یاد داشت که یافتههای اولیه آزمایشگاهی معمول که در بیماران فونویلبراند دیده میشود، به طور کلی سبب افتراق نوع ارثی و اکتسابی(vWD و AvWS) نمیشود. بنابراین موقعی که یافتههای آزمایشگاهی احتمال تشخیص بیماری فونویلبراند را بدهند، به ویژه در حضور اختلالات زمینه مرتبط با AvWS، باید این بیماری مد نظر قرار گیرد. حتی اگر یک اختلال در میان اختلالات شناسایی شده مرتبط با AvWS نباشد، باید مورد بررسی بیشتر قرار گیرد چرا که ممکن است این اختلال جدید مرتبط با AvWS بوده و قبلاً شناسایی نشده باشد(28).
با این حال، تشخیص افتراقی AvWS و vWD ارثی وابسته به نوع بیماری زمینه ایجادکننده AvWS است. مثلاً در بیماران MPDs ، بررسیهای بافتشناسی و گستره مغز استخوان میتواند کمککننده باشد. در بیماران قلبی - عروقی، نشانههای خونریزی شک تشخیص را متوجه AvWS میکند، و یا در بیماران بدون سابقه یا نشانههایی از بیماریهای مرتبط با ایجاد AvWS میتوان به دنبال سابقه مصرف دارو گشت(95).
در واقع دو گروه اصلی از بیماران وجود دارند که در آنها تشخیص AvWS باید مورد توجه باشد، یکی بیماران خونریزیدهنده که در آنها آزمونهای آزمایشگاهی بیانگر احتمال وجود اختلال مرتبط با vWF باشند و دوم، بیماران با اختلالات شناختهشده مرتبط با AvWS که به منظور اعمال جراحی و یا روشهایی که با خطر بالای خونریزی همراه هستند، مشاوره میگیرند(28).
متداولتریـن آزمـونهای مـورد استفاده در تشخیص و
افتراق نوع ارثی و اکتسابی بیماری فونویلبراند از آزمونهای اولیه و عمومی تشخیصی تا آزمونهای تخصصی را در بر میگیرد.
1-4 آزمونهای روتین و عمومی:
در واقع آزمونهای اولیه مورد استفاده در تشخیص AvWS همانهایی هستند که برای vWD ارثی به کار میروند(28). یک مطالعه گروهی انجام شده توسط تیده و همکاران در سال 2008 نشان داد که BT، PT و aPTT برای تشخیص AvWS مفید نیست(30). آزمونهای سنجش فاکتور 8 انعقادی(FVIII:C)، آنتیژن فونویلبراند (vWF:Ag)، کوفاکتور ریستوستین(vWF:RCo)، و فعالیت اتصالیvWF به کلاژن(collagen binding capacity, vWF:CB) در برخی مواقع بیماری و اغلب در LPDs کاهش یافته هستند(30، 29).
نقص در هموستاز اولیه با طولانی شدن زمان خونریزی پوستی(BT) یا زمان انسداد (Closure time, CT) طولانی با استفاده از دستگاه PFA-100 مشخص میشود. سطح vWF:Ag پلاسمایی طبیعی یا با کاهش خفیف بوده که در تضاد با کاهش محسوستر vWF:RCo و vWF:CB است. در واقع سنجشهای فعالیت کیفیvWF (از جمله vWF:RCo و vWF:CB) اغلب خوانش پایینتر نسبت به سنجش کمّی vWF:Ag دارند. در نتیجه نسبت vWF:RCo/vWF:Ag اغلب کمتر از 7/0 و شبیه به نوع A2 vWD است. در زمانی که سطح FVIII:C نیز پایین باشد، aPTT نیز طولانی میشود(96).
چندین شاخص(index) آزمایشگاهی، فعالیت، آنتیژن و منومرهای vWF را ارزیابی میکنند. ارزیابی vWF:Ag، مونومر vWF را صرف نظر از پلیمریزاسیون آن اندازهگیری میکند. vWF:RCo ظرفیت اتصالی vWF را به گلیکوپروتئین Ib پلاکتی اندازهگیری میکند. بر خلاف آزمونهای سنجش آنتیژن، vWF:RCo تا حدودی به یکپارچگی مولتیمری vWF وابسته است. در شرایط طبیعی، vWF:RCo با vWF:Ag همخوانی دارد و نسبت vWF:RCo/Ag نزدیک به 1 است. در AvWS، نسبت کاهش یافته vWF:RCo/Ag بیانگر وجود آنتیبادیهای مهارکننده، کاهش انتخابی و یا فقدان HMWMs میباشد. ظرفیت اتصالی به کلاژن(vWF:CB)، اتصال vWF به گیرنده کلاژن پلاکتی را اندازهگیری میکند و با آنتیژن vWF در اتباط است. نسبت کاهشیافته vWF:CB/Ag میتواند بیانگر کاهش انتخابی یا فقدان HMWMs باشد (97، 44). در مجموع، نسبت کاهشیافته vWF:RCo/Ag یا vWF:CB/Ag میتواند بیانگر اختلالات ساختاری یا کارکردی vWF باشد حتی اگر فعالیت کامل مولکول در محدوده طبیعی باشد(30).
جامعترین توصیفی که در مورد AvWS در دسترس است، از گزارش ثبتی(رجیستری) بینالمللی انجام شده توسط ISTH میباشد. در این مطالعه، 186 بیمار بر اساس سه معیار زیر وارد مطالعه شدند:
1- تاریخچه اکتسابی خونریزی 2- مقادیر پایین vWF:RCo یا vWF:CB و 3- نسبتهای vWF:RCo/Ag و vWF:CB/Ag کمتر از 7/0 در مواردی که vWF:RCo و vWF:CB لب مرزی یا طبیعی هستند(29). سپس در کمیته ISTH در سال 2008 معیارهای فوق به عنوان الگوریتم تشخیصی ضروری برای تشخیص AvWS به کار گرفته شدند(98).
در مجموع، الگوریتمهای تشخیصی استفاده از سنجش فاکتور 8 (FVIII:C)، vWF:Ag، vWF:RCo، و vWF:CB را پیشنهاد میکنند، اما حساسیت این سنجشها و دیگر آزمونهای آزمایشگاهی برای تشخیص AvWS به خوبی مشخص نیست. به علاوه اطلاعات کمی درباره قابلیت آزمونهای دیگر از جمله آزمونهای تشخیصی بر بالین بیمار یا آزمونهای نقطه مراقبت(point of care testing, POCT) از جمله PFA-100 در دسترس است. پیشنهاد شده که این آزمون به عنوان بخشی از ورکاپ تشخیصی بیماری انجام شود، اما نتایج متضادی درباره حساسیت آن گزارش شده است. نسبت کاهشیافته vWF:RCo/Ag یا vWF:CB/Ag میتواند بیانگر اختلالات ساختاری یا کارکردی vWF باشد حتی اگر فعالیت کامل مولکول در محدودهی طبیعی باشد(30).
لازم به یادآوری است که هیچکدام از آزمونهای عمومی ذکر شده در بالا تمایزی بین AvWS و نوع ارثی vWD ایجاد نمیکنند.
2-4 پروپپتید vWF :
در مطالعههای گزارش شـده کـه اندازهگیـری سطـوح
پروپپتید vWF (vWF-PP) به دلیل این که به عنوان یک نشانگر بیوسنتز vWF مطرح است و افزایش پاکسازی vWF را از جریان خون منعکس میکند، تشخیص AvWS را بهبود میبخشد. نسبت افزایشیافته vWF:PP/vWF:Ag که در حالت عادی 1:1 است، بیانگر افزایش پاکسازی vWF از پلاسما است. با این حال، این افزایش در زیرگروهی از بیماران تیپ vWD I هم دیده میشود. بنابراین، اندازهگیری vWF:PP و نسبت vWF:PP/Ag، همیشه بین AvWS و vWD تمایز قایل نمیشود و برای استفاده معمول در تشخیص افتراقی توصیه نمیشود. همچنین AvWS اغلب با سنتز طبیعی ولی پاکسازی سریع vWF از گردش خون مشخص میشود، و در نتیجه سطوح پروپپتید اغلب طبیعی است(101-99، 24، 14).
3-4 سنجشهای مولتیمری:
الکتروفورز مولتیمر vWF با اثبات نقص مولتیمری HMW به تشخیص AvWS از نوع 1 vWD کمک میکند (102). فقدان یا کاهش HMWMs را میتوان با استفاده از تراکمسنجی(densitometry) به طور کمّی انجام داد. با این حال این روشها مشکل و زمانبر بوده و در بسیاری از آزمایشگاهها در دسترس نیستند و به علاوه این که هنوز استانداردسازی نشدهاند. افزون بر این، متغیرهای پیشسنجهای(preanalytical) میتوانند سبب از دست رفتن کاذب HMWMs شوند (103، 28). طبق گزارشها، استاندارد طلایی برای شناسایی ناهنجاریهای ساختاری vWF، سنجش مولتیمری با استفاده از جداسازی الکترفورزی و رنگآمیزی ایمنی (immunostaining) است(97). موقعی که سنجش مولتیمری به درستی به کار رود، میتواند به عنوان یک ابزار حساس برای شناسایی ناهنجاریهای ساختاری vWF مطرح باشد و الگوهای تیپیک اختصاصی به دست آمده برای اختلالات مختلف میتواند در افتراق AvWS از vWD ارثی کمککننده باشد(33). کاهش HMWMs بعضی مواقع در بیماران با نتایـج طبیعـــی RCo و CB و حتـی نسبـتهای طبیـعی
در این بیماران به طور معمول، vWF:Ag، vWF:RCo و vWF:CB در سطح طبیعی هستند و یا حتی افزایش دارند. نسبتهای vWF:RCo/Ag و vWF:CB/Ag اغلب اما نه همیشه، کاهشیافته هستند. در برخی بیماران، کاهش یا فقدان HMWMs تنها ناهنجاری آزمایشگاهی نشاندهنده AvWS است. بنابراین فقدان HMWMs به تنهایی میتواند یک فاکتور خطر خونریزی در بیماران قلبی عروقی محسوب شود(54-52، 50، 48، 39). علاوه بر بیماریهای زمینهای قلبی عروقی، AvWS در دیگر بیماریهایی که با اختلالات قلبی همراه هستند مانند سندروم نونان(Noonan syndrome) نیز گزارش شده است (56، 55).
2-3 بدخیمیها:
1-2-3 اختلالات لنفوپرولیفراتیو:
اختلالات مختلف لنفوپرولیفراتیو(LPDs ، Lymphoproliferative ( مسئول بخش قابل توجهی از موارد AvWS هم در گزارشهای مختلف منتشر شده(30%) و هم در گزارش ثبتی ISTH (48%) است(57، 29). در یک مطالعه آیندهنگر توسط موهری و همکاران، 8 مورد از 113 بیمار(7%) مبتلا به LPDs، با AvWS همراه بودند که در50% بیماران خونریزی بالینی اثبات شد(58). در یک مطالعه دیگر توسط تیده و همکاران، 11 مورد از 35 بیمار AvWS مورد مطالعه (31%)، در نتیجه گاموپتی مونوکلونال از جمله میلوم متعدد (multiple myeloma) و ماکروگلوبولینمی ولدنشتروم (Waldenström’s disease) بودند که قبل از تشخیص AvWS با خونریزی تظاهر کرده بودند(30). البته باید یادآوری شود که خونریزی در نتیجه AvWS در LPDs به تنهایی رخ نمیدهد، مثلاً در بیماران مبتلا به بیماری ولدنشتروم یا IgM-MGUS (monoclonal gammopathy of undetermined significance)، خونریزی میتواند حاصل از افزایش ویسکوزیته خون باشد(62-59، 30). دیگر اختلالات LPDs مانند لوسمی مزمن لنفوسیتی (chronic lymphocytic leukemia, CLL) و لنفوم غیر هوچکین(non-Hodgkin lymphoma, NHL) و دیگر لنفومها مثل لنفوم لنفوپلاسماسیتیک (Lymphoplasmacytic Lymphoma) و آمیلوئیدوز نیز با شیوع کمتری به عنوان عامل AvWS معرفی شدهاند(66-63).
2-2-3 نئوپلاسمهای میلوپرولیفراتیو:
AvWS مرتبـط بـا نئـوپـلاسـمهـای میلوپرولیفراتیـو (myeloproliferative neoplasia, MPNs) بـه طـور اولیه در نتیجه جذب سطحی vWF به سلولهای خونی تغییر شکل یافته به ویژه پلاکتها حاصل میشود(67، 57). این موضوع در ترومبوسیتمی اساسی (essential thrombocythemia, ET) و پلیسیتمی ورا (polycythemia vera, PV)، همچنین در لوسمی مزمن میلوئیدی (chronic myelocytic leukemia, CML)، میلوفیبروز اولیه (primary myelofibrosis, PM) و برخی مواقع در لوسمیهای حاد گزارش شده است(72-68). در میان MPNs، شیوع AvWS در ET شایعتر از بقیه گزارش شده است (57).
در مطالعه موهری و همکاران، AvWS در 14 مورد از 125 بیمار مبتلا به اختلالات MPN (11%) گزارش گردید (58).
لازم به ذکر است که فاکتورهای خطر خونریزی در MPN به خوبی توصیف نشده است، اما بیشتر مطالعهها گزارش کردهاند که شمارش پلاکتی بسیار بالا(بیشتر از /μL103×1500) یک فاکتور خطر با اهمیت برای ایجاد خونریزی است(73). در دو مطالعه گزارش شده توسط ون ژندرن و همکاران در سال 1994، از بین 100 بیمار با ترومبوسیتمی هموراژیک، میانگین ± انحراف معیار شمارش پلاکتی nL/2016±1070 بود. بر همین اساس، یک ارتباط معکوس بین شمارش پلاکتی و نسبت vWF:RCo/Ag یا vWF:CB/Ag در بیماران MPN گزارش گردید(28). با این حال، در مطالعه آیندهنگر موهری و همکاران، شمارش پلاکت میانه(median) بیماران مبتلا به MPN و AvWS تنها /nL638 (با دامنه /nL120-1305) بود (58). بنابراین این موضوع به نظر محتمل میرسد که عوامل خطر دیگر مثل نقایص کارکردی پلاکتها نیز در خونریزی مشارکت دارند. با این حال، ارتباط بین شمارش پلاکتی، AvWS ، نقایص کارکردی پلاکتها و خطر خونریزی مستلزم مطالعه بیشتر است. بنابراین پیشنهاد میشود که بیماران خونریزیدهنده با MPN به منظور هر دو اختلال کارکردی پلاکتی و AvWS غربال شوند(73، 39).
3-3 دیگر اختلالات مرتبط با AvWS :
اختـلالات دیگـر مرتبـط با AvWS بسیار متعدد هستند ولی درصد کمی از علل ایجاد کننده بیماری را دربر میگیرند. هر چند نخستین گزارش AvWS در SLE گزارش شده است، اما اختلالات ایمنی و خود ایمن مثل- SLE ، آرتریت روماتوئید، اختلالات بافت همبند مانند بیماری بافت همبند مختلط(mixed connective tissue disease) و نشانگان اهلرز-دنلوس (Ehlers–Danlos syndromes)، سندروم آنتیفسفولیپید (antiphospholipid antibody syndrome)، حضور ضد انعقادهای شبه لوپوسی(lupus-like anticoagulants)، سندروم شوگرن(Sjogren's syndrome)، اسکلروز منتشر(systemic sclerosis) یا اسکلرودرما (scleroderma)، اختلال استخوانزایی(Osteogenesis Imperfecta) و اختلال دومفصلی خوشخیم(Benign Joint Hypermobility Syndrome) با شیوع کمتری به عنوان عامل ایجاد AvWS گزارش شدهاند(83-74، 49).
به غیر از اختلالات LPDs و MPNs ، بدخیمیهای دیگر مانند تومور ویلمز، ماستوسیتوز منتشر(systemic mastocytosis)، اختلالات متابولیکی و ذخیرهای مانند هیپوتیروئیدی، اورمی، بیماری گوشه و آنژیودیسپلازی دستگاه گوارش با شیوع کمتری به عنوان عامل AvWS معرفی شدهاند(90-84، 33).
به علاوه برخی از داروها از قبیل اسید والپروئیک (ضد تشنج)، نشاسته هیدروکسی اتیل(به عنوان انبساط دهنده پلاسما در موارد شوک هیپوولمیک)، برخی آنتیبیوتیکها مانند گریزئوفولوین(ضد قارچ)، تتراسایکلین، سیپروفلوکساسین و سفالوسپورین(ضد باکتری)، عوامل ترومبوتیک و استفاده از آفتکشهــا نیز به عنوان علت نادر AvWS گزارش شدهاند(93-91).
4- تشخیص بیماری:
دستیابی تشخیصی به AvWS به خاطر تظاهر بالینی متغیر و آزمونهای مختلف مورد نیاز برای اثبات یا رد تشخیص آن چالشبرانگیز و مشکل است و اساساً هیچ آزمون واحدی برای اثبات یا رد تشخیص بیماری در دسترس نیست(93، 57، 28). نشانههای بالینی خونریزی در بیماران AvWS به طور عمده همانند نوع ارثی از جمله خونریزیهای پوستی- مخاطی میباشد ولی مشخصترین یافته بالینی این است که بیماران به طور عمده تاریخچه خونریزیدهنده در خود یا خانواده خود ندارند. دیگر یافتههای بالینی بستگی به نشانههای بیماری زمینهای دارد (93). در غیاب سابقه خانوادگی و یا فردی خونریزی، آزمونهای تشخیصی برای AvWS همان آزمونهای مورد استفاده برای تشخیص vWD ارثی هستند(94).
باید به یاد داشت که یافتههای اولیه آزمایشگاهی معمول که در بیماران فونویلبراند دیده میشود، به طور کلی سبب افتراق نوع ارثی و اکتسابی(vWD و AvWS) نمیشود. بنابراین موقعی که یافتههای آزمایشگاهی احتمال تشخیص بیماری فونویلبراند را بدهند، به ویژه در حضور اختلالات زمینه مرتبط با AvWS، باید این بیماری مد نظر قرار گیرد. حتی اگر یک اختلال در میان اختلالات شناسایی شده مرتبط با AvWS نباشد، باید مورد بررسی بیشتر قرار گیرد چرا که ممکن است این اختلال جدید مرتبط با AvWS بوده و قبلاً شناسایی نشده باشد(28).
با این حال، تشخیص افتراقی AvWS و vWD ارثی وابسته به نوع بیماری زمینه ایجادکننده AvWS است. مثلاً در بیماران MPDs ، بررسیهای بافتشناسی و گستره مغز استخوان میتواند کمککننده باشد. در بیماران قلبی - عروقی، نشانههای خونریزی شک تشخیص را متوجه AvWS میکند، و یا در بیماران بدون سابقه یا نشانههایی از بیماریهای مرتبط با ایجاد AvWS میتوان به دنبال سابقه مصرف دارو گشت(95).
در واقع دو گروه اصلی از بیماران وجود دارند که در آنها تشخیص AvWS باید مورد توجه باشد، یکی بیماران خونریزیدهنده که در آنها آزمونهای آزمایشگاهی بیانگر احتمال وجود اختلال مرتبط با vWF باشند و دوم، بیماران با اختلالات شناختهشده مرتبط با AvWS که به منظور اعمال جراحی و یا روشهایی که با خطر بالای خونریزی همراه هستند، مشاوره میگیرند(28).
متداولتریـن آزمـونهای مـورد استفاده در تشخیص و
افتراق نوع ارثی و اکتسابی بیماری فونویلبراند از آزمونهای اولیه و عمومی تشخیصی تا آزمونهای تخصصی را در بر میگیرد.
1-4 آزمونهای روتین و عمومی:
در واقع آزمونهای اولیه مورد استفاده در تشخیص AvWS همانهایی هستند که برای vWD ارثی به کار میروند(28). یک مطالعه گروهی انجام شده توسط تیده و همکاران در سال 2008 نشان داد که BT، PT و aPTT برای تشخیص AvWS مفید نیست(30). آزمونهای سنجش فاکتور 8 انعقادی(FVIII:C)، آنتیژن فونویلبراند (vWF:Ag)، کوفاکتور ریستوستین(vWF:RCo)، و فعالیت اتصالیvWF به کلاژن(collagen binding capacity, vWF:CB) در برخی مواقع بیماری و اغلب در LPDs کاهش یافته هستند(30، 29).
نقص در هموستاز اولیه با طولانی شدن زمان خونریزی پوستی(BT) یا زمان انسداد (Closure time, CT) طولانی با استفاده از دستگاه PFA-100 مشخص میشود. سطح vWF:Ag پلاسمایی طبیعی یا با کاهش خفیف بوده که در تضاد با کاهش محسوستر vWF:RCo و vWF:CB است. در واقع سنجشهای فعالیت کیفیvWF (از جمله vWF:RCo و vWF:CB) اغلب خوانش پایینتر نسبت به سنجش کمّی vWF:Ag دارند. در نتیجه نسبت vWF:RCo/vWF:Ag اغلب کمتر از 7/0 و شبیه به نوع A2 vWD است. در زمانی که سطح FVIII:C نیز پایین باشد، aPTT نیز طولانی میشود(96).
چندین شاخص(index) آزمایشگاهی، فعالیت، آنتیژن و منومرهای vWF را ارزیابی میکنند. ارزیابی vWF:Ag، مونومر vWF را صرف نظر از پلیمریزاسیون آن اندازهگیری میکند. vWF:RCo ظرفیت اتصالی vWF را به گلیکوپروتئین Ib پلاکتی اندازهگیری میکند. بر خلاف آزمونهای سنجش آنتیژن، vWF:RCo تا حدودی به یکپارچگی مولتیمری vWF وابسته است. در شرایط طبیعی، vWF:RCo با vWF:Ag همخوانی دارد و نسبت vWF:RCo/Ag نزدیک به 1 است. در AvWS، نسبت کاهش یافته vWF:RCo/Ag بیانگر وجود آنتیبادیهای مهارکننده، کاهش انتخابی و یا فقدان HMWMs میباشد. ظرفیت اتصالی به کلاژن(vWF:CB)، اتصال vWF به گیرنده کلاژن پلاکتی را اندازهگیری میکند و با آنتیژن vWF در اتباط است. نسبت کاهشیافته vWF:CB/Ag میتواند بیانگر کاهش انتخابی یا فقدان HMWMs باشد (97، 44). در مجموع، نسبت کاهشیافته vWF:RCo/Ag یا vWF:CB/Ag میتواند بیانگر اختلالات ساختاری یا کارکردی vWF باشد حتی اگر فعالیت کامل مولکول در محدوده طبیعی باشد(30).
جامعترین توصیفی که در مورد AvWS در دسترس است، از گزارش ثبتی(رجیستری) بینالمللی انجام شده توسط ISTH میباشد. در این مطالعه، 186 بیمار بر اساس سه معیار زیر وارد مطالعه شدند:
1- تاریخچه اکتسابی خونریزی 2- مقادیر پایین vWF:RCo یا vWF:CB و 3- نسبتهای vWF:RCo/Ag و vWF:CB/Ag کمتر از 7/0 در مواردی که vWF:RCo و vWF:CB لب مرزی یا طبیعی هستند(29). سپس در کمیته ISTH در سال 2008 معیارهای فوق به عنوان الگوریتم تشخیصی ضروری برای تشخیص AvWS به کار گرفته شدند(98).
در مجموع، الگوریتمهای تشخیصی استفاده از سنجش فاکتور 8 (FVIII:C)، vWF:Ag، vWF:RCo، و vWF:CB را پیشنهاد میکنند، اما حساسیت این سنجشها و دیگر آزمونهای آزمایشگاهی برای تشخیص AvWS به خوبی مشخص نیست. به علاوه اطلاعات کمی درباره قابلیت آزمونهای دیگر از جمله آزمونهای تشخیصی بر بالین بیمار یا آزمونهای نقطه مراقبت(point of care testing, POCT) از جمله PFA-100 در دسترس است. پیشنهاد شده که این آزمون به عنوان بخشی از ورکاپ تشخیصی بیماری انجام شود، اما نتایج متضادی درباره حساسیت آن گزارش شده است. نسبت کاهشیافته vWF:RCo/Ag یا vWF:CB/Ag میتواند بیانگر اختلالات ساختاری یا کارکردی vWF باشد حتی اگر فعالیت کامل مولکول در محدودهی طبیعی باشد(30).
لازم به یادآوری است که هیچکدام از آزمونهای عمومی ذکر شده در بالا تمایزی بین AvWS و نوع ارثی vWD ایجاد نمیکنند.
2-4 پروپپتید vWF :
در مطالعههای گزارش شـده کـه اندازهگیـری سطـوح
پروپپتید vWF (vWF-PP) به دلیل این که به عنوان یک نشانگر بیوسنتز vWF مطرح است و افزایش پاکسازی vWF را از جریان خون منعکس میکند، تشخیص AvWS را بهبود میبخشد. نسبت افزایشیافته vWF:PP/vWF:Ag که در حالت عادی 1:1 است، بیانگر افزایش پاکسازی vWF از پلاسما است. با این حال، این افزایش در زیرگروهی از بیماران تیپ vWD I هم دیده میشود. بنابراین، اندازهگیری vWF:PP و نسبت vWF:PP/Ag، همیشه بین AvWS و vWD تمایز قایل نمیشود و برای استفاده معمول در تشخیص افتراقی توصیه نمیشود. همچنین AvWS اغلب با سنتز طبیعی ولی پاکسازی سریع vWF از گردش خون مشخص میشود، و در نتیجه سطوح پروپپتید اغلب طبیعی است(101-99، 24، 14).
3-4 سنجشهای مولتیمری:
الکتروفورز مولتیمر vWF با اثبات نقص مولتیمری HMW به تشخیص AvWS از نوع 1 vWD کمک میکند (102). فقدان یا کاهش HMWMs را میتوان با استفاده از تراکمسنجی(densitometry) به طور کمّی انجام داد. با این حال این روشها مشکل و زمانبر بوده و در بسیاری از آزمایشگاهها در دسترس نیستند و به علاوه این که هنوز استانداردسازی نشدهاند. افزون بر این، متغیرهای پیشسنجهای(preanalytical) میتوانند سبب از دست رفتن کاذب HMWMs شوند (103، 28). طبق گزارشها، استاندارد طلایی برای شناسایی ناهنجاریهای ساختاری vWF، سنجش مولتیمری با استفاده از جداسازی الکترفورزی و رنگآمیزی ایمنی (immunostaining) است(97). موقعی که سنجش مولتیمری به درستی به کار رود، میتواند به عنوان یک ابزار حساس برای شناسایی ناهنجاریهای ساختاری vWF مطرح باشد و الگوهای تیپیک اختصاصی به دست آمده برای اختلالات مختلف میتواند در افتراق AvWS از vWD ارثی کمککننده باشد(33). کاهش HMWMs بعضی مواقع در بیماران با نتایـج طبیعـــی RCo و CB و حتـی نسبـتهای طبیـعی
شکل 3: سنجش مولتیمری در بیماران فونویلبراند ارثی و اکتسابی و مقایسه با نمونه پلاسمای طبیعی(normal plasma, NP). الف- مقایسه سنجش مولتیمری در AvWS به علت بیماریهای قلبی - عروقی با vWD نوع A2 و B2. ب- AvWS به علت MGUS. ج) AvWS به علت MPDs (28).
4-4 سنجش اتوآنتیبادیها:
بر خلاف هموفیلی اکتسابی، تعداد کمی از موارد AvWS با سازوکار اتوآنتیبادی بر ضد فعالیت vWF مرتبط با پلاکت مشخص میشوند. با این وجود، به نظر میرسد حضور آنتیبادیهای نوترالیزان با تمایل شدیدتری به خونریزی همراه باشد و به همین دلیل غربال آنتیبادی باید در همه موارد AvWS انجام شود(104، 99، 93، 57).
متداولترین روش مورد استفاده برای جستجوی فعالیت بازدارنده علیه vWF بر اساس مطالعات پلاسمای مخلوط (mixing studies) پلاسمای بیمار با پلاسمای معمولی و انکوباسیون در دمای 37 درجه سانتیگراد و به دنبال آن اندازهگیری vWF:RCo و vWF:CB اضافی (residual) است. با این حال، مهارکنندهها به ندرت شناسایی میشوند و آنتیبادیهایی که به vWF متصل شده و پاکسازی پلاسمایی آن را بدون خنثی کردن فعالیت vWF تسریع میکنند، با این روش قابل شناسایی نیستند. سنجشهای الایزا نیز به عنوان یک گزینه مطرح هستند اما هنوز به اندازه کافی استانداردسازی نشدهاند(105، 99، 64، 57).
اتوآنتیبادیهای آنتی-vWF در پاتوژنز برخی بیماران AvWS به ویژه مبتلایان به اختلالات لنفوپرولیفراتیو(LPDs) نقش دارند و شناسایی آنها یک ممیزهی تشخیصی(pathognomonic) برای سازوکار اکتسابی کمبود vWF است. همچنین به نظر میرسد که حضور این اتوآنتیبادیها با افزایش تمایل به خونریزی در بیماران همراه است. با این حال این آنتیبادیها تنها در 14 درصد موارد مشکوک به AvWS شناسایی شدهاند و ویژگیهای کارکردی متنوعی را نشان میدهند. همچنین به نظر میرسد حضور اتوAbها با خونریزی شدیدتر همراه است(58، 30).
بـر خلاف هموفیلی اکتسابی که مهارکنندههای فاکتور 8 عملاً همیشه عامل بیماری میباشند و با ارزیابیهای آزمایشگاهی استاندارد قابل شناسایی هستند، تنها در تعداد کمی از بیماران AvWS میتوان Abهای خنثیکننده یا نوترالیزان را با استفاده از ارزیابیهای خنثیکنندگی vWF:Ag، vWF:RCo، vWF:CB یا RIPA و مطالعههای پلاسمای مخلوط شناسایی کرد. با این حال، این ارزیابیها از نظر تکنیکی مشکل بوده و قادر به شناسایی آنتیبادیهای غیرنوترالیزان مرتبط نیستند. بنابراین، شناسایی مهارکنندهها در درصد کمی از بیماران AvWS ضرورتاً به معنای عدم حضور اتوآنتیبادیها نیست، بلکه ممکن است منعکسکننده حضور اتوAbهای غیرنوترالیزان باشد که پاکسازی vWF را از جریان خون سرعت میبخشند بدون اینکه روی سنجههای کارکردی قابل اندازهگیری اثر مهارکنندگی داشته باشند(107، 106، 57).
افزون بر اتوآنتیبادیها ، یافتن یک پروتئین مونوکلونال توسط الکتروفورز پروتئینهای سرمی نیز برای تشخیص AvWS محل بحث است. همچنین برخی مواقع مهارکنندهها با vWF کمپلکسهای اشباع تشکیل میدهند و بنابراین مانع شناسایی خود میشوند، مگر این که با کمک حرارت یا دیگر روشهای فیزیکی یا شیمیایی کمپلکسها تجزیه شود. همچنین یک روش الایزا معرفی شده که طیف وسیعتری از آنتیبادیهای متصلشونده به vWF را شناسایی میکند. آنتیبادیها غیرنوترالیزان متصلشونده به vWF در بیماران LPDs و نیز دیگر اختلالات زمینهای گزارش شده و میتوان آنها را توسط ELISA شناسایی کرد هر چند هنوز هیچ ارزیابی استانداردسازی شدهای برای شناسایی آنها در دسترس نیست. لازم به ذکر است که vWF مشتق از پلاسما حاوی آنتیژن گروه خونی ABO است و نباید به عنوان آنتیژن برای شناسایی اتوAbها در ELISA استفاده شود. به دلیل اینکه حضور ایزوآگلوتینینها میتواند سبب نتایج مثبت کاذب گردد. با این حال، میتوان برای رفع این نقیصه از vWF انسانی نوترکیب بیانشده در سلولهای حیوانی کشتشده به عنوان ریجنت استفاده کرد(105، 30، 28).
بایـد بـه خاطر داشت که تشخیص AvWS نیاز به تغییر در همه پارامترهای گفته شده در بالا ندارد. به عنوان مثال در یک مطالعه، هیلمن و همکاران AvWS را در بیماران تحت MCS ارزیابی کردند، و تشخیص بیماری را بر مبنای سنجش مولتیمری بنا نهادند که HMWMs غایب بودند و نیز دستکم یکی از نسبتهای کارکردی vWF کاهشیافته بود(108). البته ترجیح این است که تمامی سنجشهای مربوط به فاکتور فونویلبراند از جمله vWF:Ag، vWF:RCo، vWF:CB و سنجشهای مولتیمری در بیماران مشکوک به AvWS انجام شود.
لازم بـه ذکـر اسـت که نشانههای بالینی و آزمایشگاهی مرتبط با AvWS در اختلالات مختلف به طور معمول باعث ایجاد بیماری فونویلبراند نوع 2 میشود، هر چند در برخی بیماریها و حالات مختلف، بیمـاری نــوع 1 و یا 3 هم گزارش شده است(109).
همچنین باید به یاد داشت که اختلالات مرتبط با AvWS در جمعیت به نسبت معمول هستند. بنابراین انجام آزمونهای غربالگری جمعیتی ضروری نیست و حتی میتواند منجر به کسب نتایج مثبت کاذب فراوان و در نتیجه درمانهای غیر ضروری گردد. بنابراین انجام آزمونهای تشخیصی در بیماران بیعلامت، باید بر مبنای قضاوت بالینی پزشک هر بیمار باشد(28).
5- نشانههای بالینی و مدیریت درمان بیماری:
نشانههای بالینی در نوع ارثی و اکتسابی بیماری ویلبراند به طور عمده یکسان هستند. بنابراین انجام آزمونهای غربالگری در بیماران دارای اختلالات زمینهای مرتبط با AvWS قبل از جراحی ماژور و دیگر مداخلات با خطر بالای خونریزی پیشنهاد میشود(29).
نشانههای بالینی این بیماری همانند نوع ارثی، به طور عمده با خونریزی از مخاط(غشاهای موکوسی) مجاری معدهای رودهای(گوارشی) و ادراری تناسلی همراه است. به علاوه هماتومهای بافت نرم و خونریزیهای پس از عمل جراحی نیز معمول هستند(34). با این حال، شدت نشانههای بالینی در AvWS بستگی به شدت بیماری زمینه دارد و با درمان بیماری زمینهای، این بیماری نیز از بین میرود. بنابراین پیشآگهی و نیز مدیریت درمان بیماری وابسته به پاتوفیزیولوژی زمینه مرتبط با بیماری است. با این حال، با وجود انجام مطالعههای متعدد در رابطه با پاتولوژی بیماری، همچنان مدیریت درمان در AvWS همانند تشخیص بیماری، به عنوان یک چالش برای متخصصین خون باقیمانده است. هیچ دستورالعمل مبتنی بر شواهدی برای مدیریت بیماری وجود ندارد. درمان بیماریهای زمینهای بسته به بیماری به وسیله کورتیکواستروئیدها، شیمیدرمانی، سرکوبگرهای ایمنی، رادیوتراپی، جراحی و یا جایگزینی دریچه قلبی(vavle replacement) در بیماران نقص دریچههای قلبی میتوانند منجر به بهبودی در گروهی از بیماران شوند، اما این درمانها همیشه امکانپذیر و یا موفق نیستند. در مواردی که این درمانها به سرعت مؤثر نباشد، درمان علامتی با هدف اصلاح کمبود vWF به کار میرود تا از خونریزی غیرطبیعی جلوگیری و یا آنرا درمان کند. برخی از این گزینههای درمانی مانند دسموپرسین، کنسانترههای حاوی vWF، کنسانترههای فاکتور 8 نوترکیب(فاقد vWF)، فاکتور 7 نوترکیب فعال(rFVIIa)، IV-Ig و پلاسمافرز میتوانند در این مواقع مؤثر باشند. به خاطر ساز و کارهای مختلف AvWS ، اغلب بیش از یک رویکرد درمانی برای مدیریت خونریزیهای حاد و یا به عنوان پیشگیری در هنگام استفاده از روشهای تهاجمی در بیماران لازم است(95، 28، 25، 18).
بحث
ممکن است بر اساس شواهد تصور شود که بیماری فونویلبراند اکتسابی یک اختلال ناشایع است، اما بهتر است گفته شود که AvWS در واقع یک بیماری است که در بیشتر موارد یا کم تشخیص داده شده (underdiagnosed) و یا به اشتباه به عنوان نوع ارثی شناسایی میشود (misdiagnosed). هر چند تشخیص AvWS و تمایز بین نوع ارثی و اکتسابی بیماری بحثبرانگیز و دشوار است ولی تشخیص افتراقی AvWS از vWD ارثی به ویژه اشکال خفیفتر بیماری، با توجه به تفاوت در رویکرد درمانی مهم است. برای تشخیص نهایی AvWS ، نیاز به یافتههای مختلف بالینی و آزمایشگاهی است و باید در مطالعههای جامع، همه اطلاعات از جمله ارزیابی بالینی و تاریخچه موارد گزارش شده و نیز نتایج آزمایشگاهی را در نظر گرفت(57، 33، 30، 29).
آزمونهای آزمایشگاهی برای تشخیص AvWS و vWD ارثی یکسان هستند. بنابراین در حضور یک بیماری زمینهای مرتبط با AvWS، زمانی که یافتههای آزمایشگاهی و بالینی بیمار پیشنهاد دهنده بیماری فونویلبراند باشد، باید به ناچار تشخیص AvWS مد نظر قرار گیرد. همچنین گرفتن شرح حال از بیمار برای تشخیص AvWS مهم است.
باید دقت شود که اخذ تاریخچه با جزییات و معاینه جسمی کافی باشد تا بیشتر اختلالات مرتبط تحت پوشش قرار گیرند. معاینات به ویژه در مورد بیماریهای قلبی - عروقی مانند تنگی دریچه آئورت اهمیت زیادی دارند. به همین ترتیب بیشتر اختلالات هماتولوژیک، بدخیم و خودایمن باید به طور بالینی مشخص و غربال شوند. به علاوه الکتروفورز پروتئین سرمی و شمارش خون کامل (CBC) میتواند به ترتیب به غربال و شناسایی گاموپاتیهای مونوکلونال و دیگر اختلالات هماتولوژیک کمک کنند(110، 28، 24).
نتیجهگیری
با توجه به گستردگی بیماریهای مرتبط با AvWS، لازم است هم متخصصین بالینی به ویژه هماتولوژیستها و متخصصین قلب و هم متخصصین آزمایشگاه توجه ویژهای به این بیماری داشته باشند تا موارد AvWS به درستی شناسایی و تشخیص داده شوند. بنابراین برای همه بیماران دارای یافتههای آزمایشگاهی مرسوم، اختلالات فاکتور ویلبراند(vWF) به همراه تاریخچه منفی خونریزی پیشین در بیمار و یا سابقه خانوادگی وی، غربال برای اختلالات مرتبط با AvWS توصیه میشود. بنابراین تظاهر دیرهنگام حوادث خونریزی در یک زمینه با تاریخچه منفی خانوادگی باید شک را به سمت AvWS ببرد.
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
