

جلد 17، شماره 3 - ( پاییز 1399 )
جلد 17 شماره 3 صفحات 257-242 |
برگشت به فهرست نسخه ها

![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Milani S, Yari F. Platelet Refractoriness and how it is formed. bloodj 2020; 17 (3) :242-257
URL: http://bloodjournal.ir/article-1-1325-fa.html
URL: http://bloodjournal.ir/article-1-1325-fa.html
میلانی سعیده، یاری فاطمه. مقاومت پلاکتی و چگونگی ایجاد آن. فصلنامه پژوهشی خون. 1399; 17 (3) :242-257



استادیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون
متن کامل [PDF 536 kb]
(1427 دریافت)
| چکیده (HTML) (3809 مشاهده)
مقدمه
کلیاتی راجع به مقاومت پلاکتی:
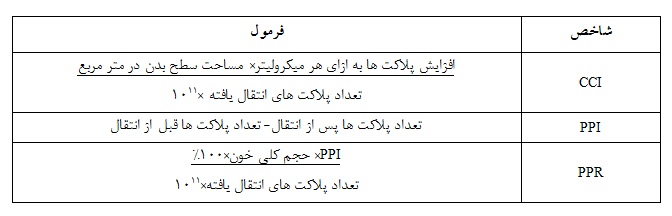
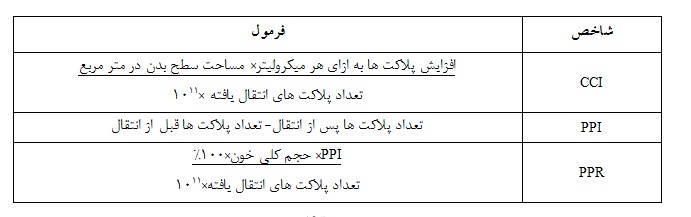
ترومبوسیتوپنی به شرایطی گفته میشود که مقدار پلاکت(ترومبوسیت) های موجود در خون، پایینتر از حد طبیعی(150000 پلاکت در هر میکرولیتر خون) باشد(1). کم بودن پلاکت در بیماران دریافتکننده مداوم این فرآورده، تحت عنوان مقاومت پلاکتی شناخته میشود. شاخصهای رایج جهت محاسبه مقاومت پلاکتی شامل افزایش تعداد تصحیح شده CCI (Corrected Count Increment)، افزایش پلاکت پس از انتقال PPI (Post-transfusion Platelet Increment) و درصد بازیافت پلاکتی PPR (Percentage Platelet Recovery) میباشند(جدول 1). با این حال محاسبه CCI یکی از بهترین پارامترها جهت بررسی وجود مقاومت پلاکتی میباشد و mL /7500CCI< پس از حداقل دو تزریق پیاپی پلاکت حاوی آنتیژنهای ABO سازگار یا تقریباً یکسان، به عنوان مقاومت پلاکتی معرفی میگردد(2).
مطالعههای مختلف نشان میدهند که سیستم ایمنی خصوصاً ایمنی همورال، مسئول 20% موارد مقاومت پلاکتی میباشد و در این بین نقش آلوآنتیبادیهای تولیدی در حذف پلاکتها از اهمیت بسیاری برخوردار میباشد(7-3). در حقیقت این آنتیبادیها، با تخریب پلاکتهای حاوی آنتیژن ناسازگار، منجر به بروز مقاومت پلاکتی میگردند. در کنار عوامل ایمونولوژیک، عوامل و فاکتورهای غیر ایمونولوژیک نیز میتوانند منجر به بروز مقاوت پلاکتی گردند(8)(جدول 2). بر خلاف عوارض ناشـی از تخریـب گلبـولهای قرمـز، تخریب پلاکتها به
صورت طبیعی، نشانههای بالینی از خود نشان نمیدهد(9).
با آشکار شدن اهمیت و نقش آنتیبادیها در فرد دریافتکننده پلاکت بر ضد آنتیژنهای بیگانه فرد اهداکننده در بروز مقاومت پلاکتی، عمده توجهات روی آنتیبادیهای ضد(Human Leukocyte Antigen) HLA متمرکز گردیده است(2). در اوایل دهه 1990، شناسایی آنتیژنهای HLA افراد دهنده و گیرنده و سازگار بودن آنها در طی تزریق پلاکت مورد توجه قرار گرفت که این امر به بهبود مقاومت پلاکتی در زیر گروهی از بیماران منجر گردید. با این حال همچنان گروه بزرگی از بیماران در خطر بروز مقاومت پلاکتی میباشند. این امر خصوصاً در مورد بیماران دارای بدخیمیهای خونی از اهمیت خاصی برخوردار میباشد(10).
گزارشهای مربوط به بروز پاسخ آلوایمنی نسبت به آنتیژنهای HLA بسیار بیشتر از آنتیژنهای HPA (Human Platelet Antigen) بوده است و به نظر میرسد آلوآنتیبادی ضد HLA ، عامل اصلی بروز مقاومتهای پلاکتی باشد. سیستم HLA از کمپلکس عمده سازگاری بافتی(MHC) منشاء میگیرد که پروتئینهای سطحی پلیمورفیک مهم جهت عرضه آنتیژن را کد میکنند. در واقع MHC یک سیستم چند شکلی و چند ژنی میباشد و حاوی لوکوسهایی برای کد کردن آلوآنتیژنهای HLA کلاس Iو II است. این ژنها نزدیک به هم و بر روی بازوی کوتاه کروموزوم 6 قرار دارند. این کمپلکس شامل 4 میلیون جفت باز DNA میباشد و کدکننده آنتیژنهایHLA-A ، Bو C (کلاس I ) و HLA-DR ، DQ و DP (کلاس II ) میباشد(12، 11).
در 12% تا 15% این بیماران که نیازمند دریافت طولانی مدت پلاکت میباشند، مقاومت پلاکتی رخ داده و افزایش ناکافی در تعداد پلاکتها به علت حذف سریع پلاکتهای انتقالی اتفاق میافتد(18). جهت درمان بیماران دارای آلوآنتیبادی ضد HLA ، تزریق پلاکتهای سازگار با HLA انجام میپذیرد. با این حال پیدا کردن پلاکتهای سازگار با HLA در مقیاس زیاد برای این بیماران با مشکل همراه است. در حال حاضر تزریق پلاکت به بیماران دارای آلوآنتیبادی بر مبنای تزریق پلاکتهای فاقد آنتیژنهای مربوط به آلوآنتیبادیهای ضد HLA در بدن این افراد استوار است با این وجود بایستی به اثرات آلوآنتی بادیهای موجود در گردش خون نیز توجه کرد(19).
در مورد سازگاری آنتیژنهای ABO ، از آنجایی که این آنتیژنها به میزان بسیار کم بر روی پلاکتها بیان گردیده و کمتر از 2 میلیلیتر گلبول قرمز در یک واحد پلاکت تزریقی وجود دارد، سازگاری ABOدر بروز مقاومت پلاکتی از اهمیت چندانی برخوردار نمیباشد. با این حال در افراد نیازمند تزریق مداوم پلاکت، میزان در معرض قرار گرفتن با گلبولهای قرمز و آنتیژنهای ABO
پلاکتی ممکن است در حدی باشد که منجر به تولید آنتیبادی در فرد دریافتکننده و همولیز پلاکتی گردد. از طرف دیگر، هر چند بیان آنتیژنهای ABO بر روی پلاکت بسیار کم بوده و وجود سازگاری ABO در تزریق پلاکت زیاد قابل توجه نمیباشد، در بیمارانی که با وجود تزریق پلاکتهای با HLA سازگار، همچنان کارآیی مؤثری در افزایش تعداد پلاکت مشاهده نمیشود، بایستی به وجود این سازگاری توجه بیشتری گردد(22-20).
ریجکرز و همکارانش نشان دادند که زیر مجموعهای از آلوآنتیبادیهای ضد HLA میتوانند باعث فعال شدن پلاکتهای اهداکنندگان و فاگوسیتوز آنها توسط ماکروفاژها گردند که در این فرآیند FcγRIIa نقش مهمی بر عهده دارد(19). مشابه این فرآیند در ترومبوسیتوپنی پورپورا ((ITP و ترانسفوزیون مرتبط با آسیب حاد ریه (TRALI) دیده شده است(24، 23). مطالعهها همچنین نشاندهنده این حقیقت است که اثرات آلوآنتیبادی بیشتر بر مبنای شناسایی اپی توپ به جای میل اتصال(افینیتی) آن استوار میباشد. همچنین هندسه اتصال آنتیبادی میتواند روی نحوه فعال شدن کمپلمان اثرگذار باشد(25).
مواد و روشها
ایمنیشناسی بروز مقاومت پلاکتی:
پلاکتهای حاوی آنتیژنهای بیگانه میتوانند عمدتاً تحت دو مسیر عمده به بدن عرضه گردند. اولین مسیر در طی بارداری بوده که یا به وسیله خونریزیهای کوچک در زمانهای مختلف بارداری اتفاق افتاده و یا عمدتاً در طی گردش خون جفتی و دوره پری ناتال پدید میآید.
آنتیژنهـای سلولهـای جنیـن به بروز پاسخ ایمنی در
مادر منجر میگردد که باعث انتقال آنتیبادیهای مادری به جنین و تخریب پلاکتهای مرتبط با این آلوآنتیبادیها میشود. این امر منجر به بروز ترومبوسیتوپنی و خونریزی در جنین و نوزاد تازه متولد شده میشود. موارد شدیدتر آن منجر به خونریزی داخل مغزی و ناتوانی شدید متعاقب آن میگردد.
ترومبوسیتوپنی آلوایمیون نوزادی- جنینی(FNAIT ؛ Foetal neonatal thrombocytopenia alloimmune)، یکی از همین موارد میباشد. بیشترین گزارشهای مربوط به پاسخ ایمنی بدن بر ضد آنتیژنهای بیگانه و تولید آلوآنتیبادی در دوران بارداری، مربوط به آنتیژنهای HLA و عمدتاً کلاس Iآن میباشد (هر چند آنتیبادی ضد HLA کلاس II نیز میتواند در طی این دوره شناسایی گردد). بر اساس روشها و یافتههایی که جهت محاسبه میزان آنتیبادیها مورد استفاده قرار میگیرند، 17% از خانمها بعد از یک بارداری، آنتیبادی ضد HLA بیان میکنند و این میزان در بارداریهای بعدی نیز افزایش مییابد(26).
دومین دلیل بروز پاسخ ایمنی ضد پلاکتها، انتقال خون میباشد که برای اولین بار به وسیله روتچر در سال 1981 ارائه گردید(27). ایجاد ایمنی ضد آنتیژنهای پلاکتی بعد از انتقال خون کامل امکانپذیر است که البته این فرآیند در کشورهای توسعهیافته بسیار نادر است. یکی از مسائلی که بایستی به آن توجه بسیاری کرد، اهدای پلاکت تازه که از زمان ذخیره آن بیش از 48 ساعت نگذشته است میباشد. چرا که در طی ذخیره پلاکت، میزان برخی مارکرها هم چون P-selectin بر روی آن افزایش مییابد که میتواند منجر به واکنش پلاکت با لکوسیتهای فرد دریافتکننده پلاکت گردد(28). همچنین در طی فرآیند ذخیره پلاکت، به علت فعالیت متابولیک پلاکت و لکوسیتهای باقیمانده، مواد غذایی مصرف گردیده و محصولات متابولیکی مضر تولید میگردند. فاکتورهای انعقادی فعال شده، بقایای سلولی و آنزیمهای پروتئولیتیک موجود در پلاسما میتوانند بر روی پلاکت تاثیر منفی بگذارند(29). از طرف دیگر انتقال سلولهای قرمز خون به علت وجود لکوسیتها که میزان زیادی آنتیژنهای HLA کلاسI را بیان میکنند و این آنتیژنها بر روی پلاکت نیز موجود میباشند، منجر به ایجاد پاسخ ایمنی ضد آنتیژنهای HLA میگردند. این امر با حذف لکوسیتها از محصولات خونی قابل پیشگیری میباشد. پرتوتابی UV نیز باعث غیر فعال شدن لکوسیتها و کاهش ایمونیزاسیون میگردد. با این حال حتی حذف لکوسیتها(حضور در محصولات پلاکتی با مقادیر کمتر از 106) نیز همچنان میتوانند ایمنیزا باشند. به طور کلی در بیماران دریافتکننده پلاکت که به طور مکرر تزریقات پلاکتی دریافت میکنند و همین طور دریافتکنندگان مکرر سلولهای قرمز خون، نوعی پان- سیتوپنی مشاهده میگردد. این افراد اگر در خطر خونریزی شدید باشند، بایستی پلاسمای درمانی نیز دریافت کنند که محصولی خونی و مملو از ایمونوگلوبولینهای واکنشی میباشد. بیمارانی که در نتیجه تزریق پلاکت، سیستم ایمنی آنها تحریک گردد ضد آنتیژنهای پلاکتی، آنتیژنهای HLA و آنتیژنهای ABH ، آنتیبادی تولید میکنند. این افراد میتوانند ضد آنتیژنهای گلبولهای قرمز هم پاسخ ایمنی از خود نشان دهند. این امر خصوصاً برای برخی از آنتیژنهای گلبولهای قرمز همچون RhD که به شدت ایمونوژن میباشند از اهمیت بیشتری برخوردار است. همچنین برخی از بیماران میتوانند در طی پیوند بافت نیز پاسخ ایمنی بر ضد آنتیژنهای HLA کلاس Iاز خود نشان دهند(30). بروز پاسخ ایمنی ضد پلاکت دریافتی، به عوامل مختلفی همچون سلولهای انتقال یافته، زمینه ژنتیکی افراد دریافتکننده پلاکت، بیماریهای زمینهای و داروهای دریافتی در این افراد(خصوصاً داروهای مهارکننده سیستم ایمنی و داروهای مداخلهکننده) بستگی دارد. عوامل دیگر همچون محیط و شرایط میکروبی نیز در این امر میتوانند تاثیرگذار باشند(30).
یافتهها
با توجه به اهمیت آلوآنتیبادی ضد آنتیژنهایHLA در بروز مقاومت پلاکتی، مطالعههای بسیاری در زمینه دلایل حساس شدن افراد گیرنده پلاکت نسبت به آنتیژنهای HLA افراد اهداکننده انجام شده است که میتواند در شناسایی مکانیسمهای دخیل در حساس شدن سیستم ایمنـی افراد دریافتکننده پلاکت نسبت به این آنتیژنها بسیار کمککننده باشد.
عمدهترین علل و مکانیسمهای حساس شدن به HLA :
1- حساس شدن به HLA در طی بارداری:
همانگونه که پیشتر گفته شد، حساسیت به HLA نتیجه عرضه آلوآنتیژنهای بیگانه به سیستم ایمنی فرد میباشد. بارداری یکی از مهمترین دلایل حساس شدن به HLA است چرا که نیمی از ژنهای جنین، بیگانه(آلوژن) میباشد و این در حالی است که سیستم ایمنی مادر، جنین در حال رشد را به لحاظ ایمنی در فضای رحم تحمل میکند اما در مقابل آنتیژنهای پدری به عنوان مثال آنتیژنهای HLA ، پاسخ ایمنی همورال و سلولار ایجاد میکند. علت وجود چنین تناقضی در پاسخهای ایمنی مادر در دوران بارداری همچنان ناشناخته باقی مانده است(31). سلولهای سایتوتروفوبلاست و سینسیشوتروفوبلاست، مرزی بین سلولهای بافت مادر و جنین ایجاد میکنند. این سلولها فاقد HLA-A و HLA-B بوده ولی HLA-G که مولکول تیپیک HLA کلاس I می باشند را ترشح میکنند. این امر جنین را از لیز شدن به وسیله سلولهای کشنده طبیعی حفظ میکند(32). این مکانیسم همراه با چند مکانیسم سازگاری دیگر، جنین را از حمله به وسیله سیستم ایمنی مادر مصون نگه میدارد. با این حال در طی رشد جفت، سلولهای سین سیشوتروفوبلاست به تدریج دچار آپوپتوز گردیده، تجزیه میگردند و محتوای سلولی خود و از جمله DNA جنینی را به جریان خون مادر آزاد میکنند. DNA جنین 7 هفته پس از بارداری در پلاسمای مادر قابل ردیابی میباشد و غلظت آن جهت ژنوتایپینگ جنین کافی است(33). بقایای این سلولهای آزاد شده میتوانند باعث بروز پاسخ ایمنی ضد HLA کلاس I گردند. هر چند سلولهای دیگری هم در این مسیر هستند که میتوانند از رحم به جریان خون مادر وارد شوند.
امروزه شکلگیری آنتیبادی ضد آنتیژن RhD در مادران فاقد این آنتیژن که جنین آنها آنتیژن RhD را روی گلبولهای قرمز خود حمل میکند به اثبات رسیده است. تیتر بالای آنتیبادیهای مادری ضد آنتیژن RhD جنین میتواند منجر به بروز بیماری همولیتیک جنینی در جنین و همین طور نوزاد تازه متولد شده گردد. به همین ترتیب، لکوسیتهای جنینی میتوانند سیستم ایمنی مادر را جهت تولید آنتیبادی ضد آنتیژنهای جنینی مانند HLA تحریک کنند. بر خلاف آنتیبادیهای مادری ضد آنتیژنهای گلبولهای قرمز همچنین آنتیژنهای گلیکوپروتئینی پلاکت و آنتیژنهای گرانولوسیتها، آنتیبادیهای مادری ضد HLA جنینی، به جنین آسیب نمیرسانند(34). میزان حساس شدن به HLA در مادران باردار بستگی به دفعات بارداری دارد و با افزایش تعداد بارداریها این میزان افزایش مییابد. همچنین روشی که برای ردیابی آنتیبادیها به کار میرود نیز در تعیین میزان حساسیت تاثیرگذار است . فاصله زمانی بررسی آنتیبادیها در زنان نسبت به زمان آخرین بارداری آنها نیز مهم میباشد و در زنانی که مدت زمان زیادی از آخرین بارداری آنها گذشته باشد، تعداد آنتیبادیهای کمتری ضد HLA قابل ردیابی خواهد بود(35).
2- حساس شدن به HLA موجود در کنسانترههای پلاکتی:
محصولات خونی مقادیر زیادی از مولکولهای HLA را به دریافتکنندگان این محصولات منتقل میکنند. گلبولهای قرمز، پلاکتها و لکوسیتهایی که ممکن است همراه این محصولات منتقل گردند، حاوی مولکولهای HLA در غشای خود میباشند. پلاسما نیز حاوی مولکولهای HLA محلول میباشد. گلبولهای قرمز منبع اصلی HLA انتقالی نبوده و تنها حدود 90 مولکول HLA بر روی سطح هر گلبول قرمز(محدوده 550-40 (موجود میباشد(37، 36). در مقابل پلاکتها حامل تعداد زیادی مولکول HLA هستند(حدود 120000-50000 مولکول HLA به ازای هر پلاکت)، هر چند که سطح پلاکتها نسبت به سطح گلبولهای قرمز بسیار کوچکتر است(38). پلاکتها تنها مولکولهای کلاس I عمدتاً HLA-A و HLA-B را بیان میکنند(40، 39). برخی یافتهها حاکی از آن هستند که پلاکتها مولکولهای HLA رها شده به وسیله سایر سلولها به پلاسما را نیز جذب میکنند(41). در یکی از مطالعههای اخیر، HLA-A*0201 که پپتیدهای مختلفی مانند پپتیدهای مشتق شده از گلیکوپروتئین IX را در ناحیه شناسایی آنتیژن ارائه میکند، در پلاکتها شناسایی شده است. HLA-A*0201که به وسیله پلاکتها بیان میگردد، در مگاکاریوسیتها یا سلولهای پیشساز آنها سرهمبندی میشود(42).
3- حساس شدن به HLA موجود در پلاسما:
مولکولهای HLA به میزان زیادی در پلاسما یافت میگردند. غلظت پروتئینهای HLA کلاس I بین 1/4-25/0 میکروگرم در هر میلیلیتر پلاسما و با وزن مولکولی 55 کیلو دالتون تخمین زده شده است(44، 43). غلظت مولکولهای HLA کلاس II ، 11 میکروگرم در هر میلیلیتر پلاسما گزارش شده است(34). در نتیجه بین 1013×1/4- 1013× 25/0 مولکول HLA کلاس I ، بیش از1010× 6/9 مولکول آلبومین و بیش از1011× 09/2 مولکول HLA کلاس I/ ایمونوگلوبولین در هر میلیلیتر از پلاسمای تازه فریز شده(Fresh Frezen Plasma )، به بیماران منتقل میگردد(45). نقش HLA محلول در ایجاد آلوآنتیبادیها زیاد مورد مطالعه قرار نگرفته است. در یک مطالعه در بیماران دچار کم کاری کلیه که تنها پلاسما دریافت کرده بودند، HLA محلول منجر به تولید آنتیبادی گردید(46). مولکول HLA کلاس I محلول میتواند با مولکول LDL ترکیب گردد و پس از فاگوسیته شدن، تجزیه و به وسیله ماکروفاژها و سایر سلولهای فاگوسیتی عرضه شود(47). لنفوسیتهای B و T میتوانند با شناسایی پپتیدهای عرضه شده ضد آنها، پاسخ ایمنی ایجاد کنند. با این حال گزارشهایی در مورد خاصیت مهارکنندگی سیستم ایمنی توسط مولکولهای HLA محلول در موش ارائه شده است. در یک مطالعه، در موشهایی که تحت درمان با HLA-B7 نوترکیب محلول قرار گرفته بودند، سیستم ایمنی همورال ضد لکوسیتهای حامل HLA-B7 ، کاملاً متوقف گردید(47).
4- حساس شدن به HLA ناشی از لکوسیتها:
لکوسیتهایی که همراه گلبولهای قرمز و کنسانترههای
پلاکتی موجود هستند، عمدهترین علت حساس شدن به HLA به شمار میروند(49، 48). لنفوسیتهای خون محیطی حدود 250000 مولکول HLA کلاس I را بر سطح خود حمل میکنند(34). همانند پلاکتها و گلبولهای قرمز، لکوسیتها هم مولکولهای HLA کلاس II را حمل میکنند. لکوسیتها میتوانند مولکولهای HLA را به پلاسمای بیماران آزاد کنند که باعث برداشت و عرضه آنها به وسیله سلولهای عرضهکننده آنتیژن(APC) میگردد. مهمتر این که لکوسیتها قادر به حمل پروتئینهای co-stimulatory میباشند. لنفوسیتهای بیماران با شناسایی مولکولهای HLAآلوژن، به آنها متصل گردیده و سیستم ایمنی آنها فعال میگردد. مولکولهای co-stimulatory روی لکوسیت افراد اهداکننده، منجر به تحریک و فعال شدن لنفوسیتهای بیماران میگردد. در نتیجه استفاده از پلاکتها و سلولهای قرمز فاقد لکوسیت، یکی از عمدهترین اقدامات جهت کاهش و جلوگیری از حساس شدن به HLA در افراد دریافتکننده پلاکت و خون میباشد(51، 50).
مقاومت پلاکتی، فرآیندی پیچیده بوده و دارای ابعاد بسیاری میباشد. مطالعههای گسترده روی FNAIT نشان داده است که مادرانی با ژنوتیپ HLA خاص، مثل DRB4*01:01 و خصوصاً DRB3*01:01 با پپتیدهای مشتق از HPA1 بیشتر واکنش میدهند تا افرادی که فاقد این ژنوتیپ هستند. نتایج مشابهی در مورد پاسخ ایمنی ضد آنتیژنهای گلبولهای قرمز و آنتیژنهای عرضه شده ناشی از پیوند بافت به دست آمده است و ژنوتیپهای خاص HLA (و به صورت کمتر برخی از آنتیژنهای سازگاری MICA و MICB) با پاسخهای ایمنی قویتر به آنتیژنهای فرد اهداکننده همراه بودهاند(53، 52).
مکانیسم عمل آلوآنتیبادیهای ضد HLA در بروز مقاومت پلاکتی:
مطالعههای قبلی نشان دادهاند که زیر گروهی از آلوآنتیبادیهای ضد HLA و سرم بیماران دارای این آنتیبادی، باعث فعال شدن پلاکت وابسته به FcgRIIa و افزایش فاگوسیتوز آنها به وسیله ماکروفاژها میگردند. اما این که چه حجم از این فعال شدن منجر به حذف پلاکتها میشود، همچنان نامشخص باقی مانده است(54). فعال شدن پلاکتها به آزاد شدن آلفا گرانولها ، ظهور (CD62P) P-selectin در سطح پلاکت و شروع شکلگیری کمپلمان به وسیله C3b ، منجر میگردد. C3b مستقیماً به P-selectin موجود بر سطح پلاکت متصل شده و فعال شدن کمپلمان منجر به جایگیری آن روی پلاکت میگردد. در این شرایط مسیر آلترناتیو کمپلمان آغاز گردیده که به دنبال آن اتصال IgG و نشست متعاقب C1q صورت میگیرد. در مرحله بعد، اتصال C3b فعال شدن کمپلمان را تسهیل نموده که در نهایت منجر به شکلگیری کمپلکس MAC (membrane attack complex) که نام دیگر آن کمپلکس C5b-9 است میگردد(55). فعال شدن کمپلمان میتواند به فعال شدن آن در فاز مایع نیز منجر گردد که در آن کندروتین سولفات آزاد شده از پلاکتهای فعال شده هدف قرار میگیرند(56). از طرف دیگر اتصال C3 به پلاکتهای فعال باعث القای میانکنش بین لکوسیت و پلاکت میگردد(57). تجمعات IgG باعث تجمع پلاکتها میگردد که این فرآیند با اضافه شدن C1q تشدید میشود (58). اضافه کردن آنتیبادیهای مونوکلونال ضد بتا-2-میکروگلوبولین و Pan HLA در مقادیر زیاد، باعث اتصال C3b و سمیت وابسته به کمپلمان(CDC) در پلاکت میگردد(60، 59).
نقش آنتیژنهای پلاکتی(HPA) در بروز مقاومت پلاکتی:
همان طور که پیش تر نیز به آن اشاره شد، یکی از دلایل ایجاد مقاومت پلاکتی، تولید آلوآنتیبادی ضد آنتیژنهای پلاکتی میباشد. یکی از دلایل ایجاد آلوآنتیبادی ضد آنتیژنهای پلاکتی، وجود پلیمورفیسم در این آنتیژنها است. این پلیمورفیسمها عمدتاً به علت تغییرات تک آمینو اسیدی در گلیکوپروتئین موجود بر سطح پلاکتها، مانند اینتگرین GPIIB/IIIA و GPIA/IIA، GPIB/V/IX تحت عنوان رسپتور فاکتور ون ویلبرند، GPIV تحت عنوان رسپتور کلاژن و ترومبواسپوندین و گلیکوپروتئین GPI CD109 میباشد(61). تفاوت زیادی در پلیمورفیسمهای HPA در جمعیتهای مختلف وجود دارد و بیماران در طی بارداری یا تزریق پلاکت، در مقابل این آنتیژنها پاسخهای آلوایمنی ایجاد میکنند. شیوع آنتیبادیهای اختصاصی ضد پلاکتها بین 11%-2% بوده و این در حالی است که حذف یا کاهش لکوسیتها، روی میزان آنها بیتاثیر میباشد(62). هر چند این آنتیبادیها شیوع فراوانی نداشته و به لحاظ آماری باعث کاهش معناداری در CCI نمیگردد. بر خلاف ترومبوسیتوپنی آلوایمیون یا پورپورای پس از انتقال، که حضور آنتیبادی Anti-HPA-1a در آن غالب میباشد، آلوآنتیبادی تولیدی در بیماران دریافتکننده پلاکت، اختصاصاً آنتیژنهای HPA-5b و HPA-1b را شناسایی میکند(63). جالب این که 70% آنتیبادیهای اختصاصی پلاکت در دورهای از عفونت در فرد ظاهر شده و ناپایدار میباشند. همچنین 50% این آنتیبادیها مانند اتوآنتیبادیها عمل میکنند(64).
زیر لایههای تنظیمی ایجاد آلوآنتیبادی:
مکانیسمهای دقیق دخیل در ایجاد ایمنی در افراد مبتلا به مقاومت پلاکتی همچنان ناشناخته باقی مانده است چرا که تعداد زیادی از افراد دریافتکننده پلاکت و مادران با بارداریهای متعدد، پاسخهای ایمنی ایجاد نمیکنند. این امر میتواند دلایل مختلفی داشته باشد. پلاکتهای بیگانه (همچنین سلولهای قرمز و لکوسیتها) انواعی از آنتیژنهای کوچک و بزرگ را بر سطح خود ارائه میکنند که برخی به عنوان واریانتهای آنتیژنیک شناخته شده و برخی به عنوان الگوهای مولکولی مرتبط با پاتوژن PAMPs
به شمار میروند.
PAMPs به عنوان، نشانههای خطر بیولوژیکی غیر خودی باعث فعال شدن سلولهای فاگوسیتی، دندریتی و لنفوسیتهایB حساس شده میگردند. این امر باعث فعال شدن پاسخهای پیش التهابی ایمنی ذاتی شده که برای عرضه آنتیژن و فعال کردن لنفوسیتهای T ضروری است. پلاکتهایی که در هنگام دریافت سیگنالهای خطر در تماس با لکوسیتها فعال میشوند، مستعد ترشح مقادیر زیادی از مولکولهای پیش التهابی هستند که در مجموع تحت عنوان Biological Response Modifiers (BRMs) به شمار میروند(66، 65). برخی از این فاکتورها و فاکتورهای ناشناخته دیگر میتوانند با سلولهای دخیل در سیستم ایمنی ذاتی، عرضه آنتیژن و پاسخهای ایمنی اکتسابی تداخل نمایند. یکی از این BRM هایی که در مقادیر زیاد به وسیله پلاکتها تولید میگردد، CD40L میباشد که در حالت محلول sCD40L و متصل به غشاء میتوانند با سلولهای عرضهکننده آنتیژن دارای CD40 مانند لنفوسیتهای B متصل گردد(67). در مدلی آزمایشی، کشت همزمان پلاکتها و لنفوسیتهای B باعث فعال شدن آنها(افزایش بیان P-selectin روی پلاکت و CD86 روی لنفوسیت B) گردید. در حقیقت میانکنش لنفوسیتهای Bو پلاکتها با تغییر در بیان CD40 و CD40L به وسیله لنفوسیتهای B و پلاکتها شناخته میشود. سه روز پس از انکوباسیون لنفوسیتها با پلاکتها، لنفوسیتهای B تمایز یافته تولید IgG1 ،IgG2 و IgG3 (اما نه IgG4 ، IgM و IgA) را افزایش میدهند(68). همچنین پاسخ لنفوسیتهای B فراخوانی شده به دلیل فعال شدن همزمان به وسیله(PRRs ؛ Pathogen Recognition Receptors) و رسپتور سلول B برای آنتیژن تسهیل میشود. اتصال آنتیژن به وسیله آنتیبادی سبب تقویت لنفوسیتهای Bواکنشدهنده به وسیله رسپتورهای FC و رسپتورهای کمپلمان میگردد. از طرف دیگر ارائه آنتیژن جهت عرضه آن به لنفوسیتهای T واکنشدهنده مهم است تا این لنفوسیتها به عنوان عملگرهای کمکی، منجر به تداوم تمایز لنفوسیتهای B واکنشدهنده گردند. شناسایی آلوآنتیژنها میتواند به وسیله دو مکانیسم صورت پذیرد: نخست شناسایی مستقیم به وسیله رسپتورهای لنفوسیتهای T ضد پپتیدهای مشتق از HLA که به وسیله سلولهای عرضهکننده آنتیژن فرد اهداکننده صورت میگیرد که این فرآیند در هنگام حذف دقیق لکوسیتها و حذف APCهای بیگانه تا حدود زیادی قابل جلوگیری میباشد. دوم پپتیدهای محلول مشتق از HLA فرد اهداکننده که به APC های فرد گیرنده متصل میگردد. آزمایش بلوکه کردن تماس APC و لنفوسیتهای T واکنشدهنده با اتصال متقاطع CTLA4-Ig باعث مهار ایجاد پاسخ ایمنی میگردد(70، 69).
ساریـس و همکارانـش نشـان دادنـد کــه پلاکتهای
اندوسیتوز شده به وسیله سلولهای دندریتی، احتمالاً به وسیله مکانیسم آپوپتوز، منجر به تحریک تولید اینترفرون گاما به وسیله لنفوسیتهای T CD4+ میگردند. این مسأله نشان میدهد که نه تنها HLA مربوط به لکوسیتهای باقیمانده در محصولات خونی بلکه HLA کلاس I پلاکتهای بیگانه هم میتوانند ایمنیزا باشند(71).
مولکولهای MHC کلاس I پلاکتی:
در گردش خون، پلاکتها بیشترین سهم را از مولکولهای MHC کلاس I در گردش به خود اختصاص میدهند و مطالعهها نشان میدهند که حدود دو سوم این مولکولهای پلاکتی، به وسیله پلاسما جذب میشوند(72). این مولکولهای MHC جذب شده از برشهای پروتئولیتیک غشای سلولهای سوماتیک حاصل میگردند. به نظر میرسد بیشتر آنتیژنهای MHC موجود بر روی پلاکتها، عمدتاً حاوی زنجیرههای سنگین و بتا-2-میکروگلوبولین باشند(73). در نتیجه بیشتر مولکولهای MHC پلاکت از نظر ساختاری تغییر یافته و توانایی جدا شدن از غشای پلاکت به دنبال ذخیرهسازی پلاکت را دارا هستند. پیشنهاد میشود که چون مولکولهای محلول MHC به محض ذخیره افزایش مییابد و ممکن است این مولکولهای مشتق از پلاکت، مسئول واکنشهای ایمنی بعدی پس از تزریق پلاکت باشند. همچنین میتواند دلیل این باشد که چرا این مولکولهای MHC محرک ضعیف لنفوسیتهای T CD8+ هستند و باعث مهار این لنفوسیتها و افزایش مقاومت در پیوندهای پوستی آلوژن میگردند(42). با این وجود گرچه بیشتر MHC های پلاکتی تجزیه میگردند این مولکولها هنوز دارای توانایی بالقوه در تحریک تولید آلوآنتیبادی میباشند. با تزریق پلاکتهای آلوژن به گیرنده، میزبان در معرض مقادیر زیادی مولکولهای تغییر یافته MHC کلاس I فرد اهداکننده قرار میگیرد. مولکولهای MHC فرد اهداکننده، سرانجام در طی مسیر گردش در طحال فاگوسیته میشوند (مثلاً کلاس I مرتبط با پلاکت) یا از طریق پینوسیتوز (کلاس I محلول) به وسیله سلولهای(مثلاً ماکروفاژها) سیستم رتیکولواندوتلیال گرفته میشوند. این مکانیسم جذب اولیه ماکروفاژهای گیرنده طحال و سلولهای دندریتی، اجازه میدهد تا به عنوان سلولهای عرضهکننده آنتیژن جهت تحریک سیستم ایمنی اکتسابی و در نهایت تولید آنتیبادی ضد آنتیژنهای فرد اهداکننده عمل کنند(73).
تولید آلوآنتیبادی ضد پلاکتهای فاقد لکوسیت:
جهت تولید آلوآنتیبادی ضد آنتیژنهای پلاکتهای انتقالی، شناسایی غیر مستقیم آلوآنتیژنها در طحال نقش کلیدی بر عهده دارد(75، 74). APC های گیرنده در طحال ابتدا بایستی پلاکتهای آلوژن را جذب کرده و آنها را به نواحی اندوزومی پردازش آنتیژن، جهت برش پروتئولیتیک MHC های پلاکتی به پپتیدهای 10-5 آمینواسیدی منتقل کنند. پپتیدها سپس به شکاف MHC II متصل میشوند و از آنجا به سطح APC جهت ارائه لنفوسیتهای CD4+ T منتقل میگردند(76). همان طور که سلولهای T از طحال عبور میکنند، گیرندههای اختصاصی پپتید-MHC آنها، آنتیژنهای موجود بر سطح APC را بررسی کرده و اگر رسپتور لنفوسیت T تمایل کافی برای اتصال بهMHC - پپتید پلاکتی(سیگنال اول) داشته باشد و شرایط CO-Stimulatory (سیگنال دوم) هم موجود باشد، لنفوسیتهای T فعال شده و به سلول افکتوری تمایز مییابند(76). سایتوکاینهای ترشح شده از سلولهای T کمکی فعال شده، با تحریک لنفوسیتهای B هدفگیری شده ضد MHC کلاس I افراد دهنده، باعث تمایز آنها به پلاسما سلها میگردد که این امر به ترشح آنتیبادی IgG ، اتصال بعدی این آنتیبادیها به پلاکت فرد اهداکننده و تخریب آنها منجر میگردد. مطالعههای حیوانی نشان دادهاند که تزریق پلاکتهای آلوژن، حالت پیش التهابی را موجب گردیده و سایتوکاینهای مربوط به لنفوسیتهای T خصوصاً لنفوسیتهای T کمکی Th1 باعث تولید اینترفرون گاما میگردد(77).
بروز مقاومت پلاکتی ناشی از عوامل غیر ایمونولوژیک:
بـرخی شرایـط کلینیکـی مـیتواند نیـاز به پلاکتها را
افزایش داده یا به تخریب آنها منجر گردد. بر اساس مطالعههایی بر روی بیماران مبتلا به لوسمی میلوئیدی حاد یا بیمارانی که تحت پیوند سلولهای بنیادی خونساز قرار گرفتهاند، شرایطی همچون تب، سپسیس، بیماری پیوند در مقابل میزبان(GVHD)، انعقاد داخل عروقی منتشر، بزرگ شدن طحال و مصرف برخی داروها میتوانند منجر به مقاومت پلاکتی گردند. تزریق پلاکت در این بیماران ممکن است با افزایش کافی تعداد پلاکت همراه نباشد. مقاومت زمانی آشکار میگردد که حداقل پس از 2 انتقال پیاپی پلاکتهای تازه(به محض تولید یا نهایتاً 2 الی 3 روز پس از تولید) با آنتیژنهای ABO سازگار، تعداد پلاکتها در بیمار به تعداد قابل قبول نباشد. توافقی بر سر این که تعداد پلاکتها بایستی 1 ساعت پس از انتقال یا 24-20 ساعت پس از انتقال محاسبه گردد وجود ندارد. همچنین فرمولی برای تصحیح حجم خون بیماران و تعداد دفعات تزریق پلاکت در این شرایط وجود ندارد. با این حال یک راهکار کلینیکی ساده جهت شناسایی بروز مقاومت پلاکتی، بررسی افزایش کمتر از 109×10 پلاکت در یک لیتر خون در بازه زمانی 24-20 ساعت پس از تزریق پلاکت میباشد(78).
سپسیس:
سپسیس به عنوان پاسخ بدن به عوامل تهدیدکننده حیات آن گفته میشود که میتواند منجر به آسیب بافت، نارسایی در عملکرد اندامهای بدن و مرگ گردد. ارتباط بین سپسیس و ترومبوسیتوپنی به خوبی آشکار شده است هر چند علت این ارتباط به خوبی مشخص نمیباشد. هماتوفاگوسیتوز که در مغز استخوان بیماران دارای سپسیس و ترومبوسیتوپنی زیاد دیده میشود، میتواند یکی از فاکتورهای دخیل در بروز مقاومت پلاکتی ناشی از سپسیس باشد(79).
همچنین کاهش تولید پلاکت نیز در برخی بیماران مبتلا
به سپسیس میتواند دلیل بروز ترومبوسیتوپنی باشد. در نهایت میتوانند در سطح اندوتلیوم فعال شده متوقف گردند که نقش مهمی در پاسخ میزبان به سپسیس بر عهده دارد. علاوه بر هموستازی، پلاکتها نقش مهمی در ایمنی ذاتی و اکتسابی داشته و پلاکتها میتوانند با لکوسیتها و سلولهای اندوتلیومی میانکنش داشته و این میانکنش باعث سوق داده شدن لکوسیتها به سمت بافت ملتهب میگردد. میانکنش اولیه پلاکت و لکوسیتها با اتصال P-selectin روی سطح پلاکت با PSGL-1 روی لکوسیت شکل میگیرد. مهار فعل و انفعالات بین پلاکت ـ نوتروفیل میتواند آسیب ریوی حاد ناشی از سپسیس را نیز بهبود بخشد(80).
پلاکتها با بیان Toll-like receptor ها که ساختارهای مولکولی روی سطح عامل پاتوژن را شناسایی میکنند، باعث ایجاد پاسخهای پیش التهابی میگردند. این فرآیند میتواند منجر به ترومبوسیتوپنی نیز بشود(81). علاوه بر این، محصولات باکتریایی مانند لیپوپلی ساکاریدها میتوانند در تعامل با آنتیبادیهای پلاکتی باعث افزایش فاگوسیتوز گردند(82). این یافتهها نشان میدهند که چگونه فاکتورهای کلینیکی مانند سپسیس میتواند منجر به مقاومت پلاکتی در افراد دریافتکننده پلاکتهای دارای آلوآنتیژن گردد.
بزرگ شدن طحال:
طحال مهمترین عامل مؤثر بر CCI پلاکتی در فرآیند انتقال میباشد. در واقع طحال به عنوان یکی از مکانهای به دام انداختن پلاکت محسوب شده و طحالهای بزرگتر حاوی ذخایر پلاکتی بیشتری هم هستند. پلاکت بزرگ باعث کاهش فواصل زمانی تزریق پلاکت میشود. مطالعهها نشان دادهاند که طحال جایگاه عمده تخریب پلاکتی میباشد. بیش از 85% پلاکتها در طحال بیماران مبتلا به اسپلنومگالی تخریب میگردند اما این میزان در گروه کنترل طبیعی 61% میباشد.
در بیماران فاقد طحال، کبد محل اصلی تخریب پلاکت محسوب میگردد که 89% تخریب در آنجا صورت میگیرد. میزان بازیافت پلاکت در گردش اندکی پس از تزریق، 26% در بیماران با طحال بزرگ، 59% در گروه کنترل و 8/97% در بیماران فاقد طحال گزارش شده است. 30 دقیقه پس از انتقال، 80% پلاکتها در طحال بیماران مبتلا بـه طحـال بـزرگ مشاهـده گردیـده اسـت در حالـی که ایـن میـزان در طحـال افـراد طبیعـی حـدود 40% بـوده
است(83).
تب:
در بیماران مبتلا به بدخیمیهای خونی، تب عامل عمده ایجاد مقاومت پلاکتی میباشند. اگر چه بروز تب با کاهش CCI همراه است. معلوم نیست حضور عوامل ایجاد کننده پیچیدگی مانند عفونت یا داروها در این امر دخیل میباشد یا خیر. بررسی بیمارانی که ضد HLA فرد اهداکننده پاسخ ایمنی ایجاد کرده بودند نشان داد که حضور تب در هنگام تزریق پلاکت به اهداکنندگان، باعث کاهش PPR (Percent Platelet Recovery ) میگردد. با این حال نکته حائز اهمیت این مطلب زمانی آشکار شد که با استفاده از پلاکتهای سازگار، تب باعث اثر مشخصی روی PPR نگردید(84).
انعقاد درون عروقی منتشر (Disseminated intravascular coagulation):
مصرف فاکتورهای انعقادی و تولید نامنظم و بیش از حد ترومبین، باعث رسوب فیبرین در عروق کوچک میشود که به این فرآیند انعقاد درون عروقی منتشر یا DIC (Disseminated Intravascular Coagulation) گویند(85). بیماران مبتلا به لوسمی پرومیلوسیتیک حاد به علت رها شدن فاکتورهای بافتی از گرانولهای سلولهای لوسمی که یا به صورت خود به خودی یا در نتیجه شیمی درمانی اتفاق میافتد، DIC از خود نشان میدهند(85).DIC میتواند منجر به غیر فعال شدن و بیحرکت گردیدن پلاکتها شده و یکی از عوامل CCI ضعیف پس از تزریق پلاکت به شمار میرود(86).
سن پلاکت:
مطالعهها نشان میدهند سن پلاکت به طرز معناداری بر
روی CCI اثر گذار است. جالب این جـا اسـت که در یک مطالعه انجام شده بر روی بیماران دارای عفونت، انعقاد درون عروقی منتشر و بزرگی طحال، مقاومت نسبت به تزریق پلاکتهای کهنه در این بیماران ایجاد گردید در حالی که این فرآیند به هنگام تزریق پلاکتهای تازه در این افراد مشاهده نشد. یکی از دلایل این امر میتواند فعالشدن پلاکتهای قدیمی در طی چرخه ذخیرهسازی باشد. در نتیجه بایستی به سن پلاکتها به عنوان یک فاکتور غیر ایمونولوژیک در بروز مقاومت پلاکتی توجه کرد(87).
داروها:
ترومبوسیتوپنی ناشی از داروها شایع میباشد و داروهای مختلفی در این رابطه شناسایی گردیدهاند که از این میان میتوان به ونکومایسین، آمفوتریسین ب و هپارین اشاره کرد(88). داروها با مکانیسم ایمنی منجر به ترومبوسیتوپنی میگردند(89).
بیماری Venoocclusive و پیوند در مقابل میزبان، Graft Versus Host Disease (GVHD ):
بحثهای فراوانی راجع به این که پیوند سلولهای بنیادی خونساز عامل خطری برای مقاومت پلاکتی محسوب میگردد یا خیر وجود دارد. انسداد عروق کوچک کبدی(VOD: Veno Occlusive Disease) کبدی در 22% بیمارانی که پیوند سلولهای بنیادی خونساز دریافت کردهاند گزارش شده است. مقاومت پلاکتی میتواند یکی از علائم وخامت کلینیکی حال بیمارانی باشد که پیوند سلولهای بنیادی خونساز دریافت کردهاند(90). GVHD نیز فاکتور خطری برای بروز مقاومت پلاکتی در بیمارانی است که پیوند سلولهای بنیادی خونساز دریافت کردهاند. میزان بروز اتوآنتیبادیهای پلاکتی در بیماران با GVHD حاد یا مزمن به طرز معناداری بالاتر از گروه کنترل میباشد(92، 91).
التهاب و مقاومت پلاکتی:
پلاکتهـا قادر به تولید مدیاتورهای التهابی مثل IL-1a،
IL-8 ، RANTES ،TGFβ و همین طور CD154 هستند(93). CD154 های مشتق از پلاکت مستقیما قادر به تحریک لنفوسیتهای B جهت تکثیر و تولید آنتیبادی میباشند. در نتیجه حتی در صورت عدم حضور لنفوسیتهای T CD4+، پلاکتها میتوانند در القای تولید آلوآنتیبادی نقش کمکی داشته باشند(94).
برخـی مطالعههـا نشـان دادهاند که شرایط التهابی بیمار، زیر گروههای APC مصرفکننده گلبولهای قرمز را تغییر داده و منجر به بروز پاسخ ایمنی و تولید آلوآنتیبادی میگردند. اثر مشابهی ممکن است پس از تزریق پلاکت اتفاق بیفتد. بیماران دریافتکننده پلاکت معمولاً دارای آسیبهای قابل توجه همچون تروما، عفونت یا بیماری میباشند. مطالعه بر روی موشهایی که به لحاظ ژنتیکی یکسان بودند نشان داد که تفاوتهای محیطی هر حیوان مانند زمینه التهابی آن میتواند روی پاسخ ایمنی آنها مؤثر باشد(96، 95).
سایر عوامل:
برخی از مطالعهها حاکی از پایین بودن مقدار پلاکت تزریقی، پایین بودن کیفیت پلاکتهای تزریق شده و اندازه (size) بدن بیمار دریافتکننده پلاکت در بروز پاسخ ایمنی میباشند(97).
نتیجهگیری
علیرغم اقدامات جدی، مقاومت پلاکتی همچنان یک عارضه مهم در بحث انتقال خون و فرآوردههای خونی بوده که هر چند در گروه کوچکی از افراد مشاهده میگردد، اما پیشگیری از این عارضه در بیمارانی که نیازمند دریافت مداوم پلاکت میباشند همچنان امری مهم و حیاتی به نظر میرسد. در این راستا شناسایی عوامل ایجاد کننده مقاومت پلاکتی در جلوگیری از بروز آن از اهمیت به سزایی برخوردار میباشد. همان گونه که گفته شد، 80% موارد مقاومت پلاکتی ناشی از عوامل غیر ایمنی و 20% آن ناشی از عوامل ایمنی و در نتیجه تولید آنتیبادی ضد آنتیژنهای موجود بر سطح پلاکت میباشد. این آنتیبادیها میتوانند آنتیبادیهای خنثیکننده تولید شده ضد آنتیژنهای ABO موجود بر روی پلاکت، یا آلوآنتیبادیهایی باشند که ضد آنتیژنهای لکوسیت انسانی(HLA) کلاس I در 80% موارد، یا آنتیژنهای پلاکت انسانی(HPA) در 20% موارد بوده و منجر به تخریب پلاکتهای انتقالی گردند. یکی دیگر از راههای تولید آلوآنتیبادی ضد آنتیژنهای HLA ، پیوند بافت میباشد. در بارداری نیز، آنتیبادی ضد آنتیژنهای لکوسیت انسانی(HLA) که بر سطح سلولهای جنین آنها(به ارث رسیده از پدر) تولید میگردد میتواند در بارداریهای بعدی یا تزریق پلاکت در خانمهای دارای سابقه بارداری ایجاد مشکل کند. پلاکتها هم چنین آنتیژنهای پلاکتی(HPA) را بیان کرده که واریانتهایی از مولکولهای تجمع یافته و چسبنده میباشند و بین مادر و جنین و فرد اهداکننده و دریافتکننده متفاوت بوده که منجر به تولید آلوآنتیبادیها و مشکلات بعدی آن میگردد. تزریق پلاکت با انتقال لکوسیتها نیز همراه است که از طریق بیان کپیهای فراوان HLA کلاس I (و کلاس II بیان شده بر روی لنفوسیتهای T فعال شده، لنفوسیتهای B و سلولهای دندریتی) به تولید پاسخ ایمنی منجر میگردند. همچنین سلولهای قرمز خونی باقیمانده در کنسانترههای پلاکتی نیز باعث تولید آلوآنتیبادی ضد آنتیژنهای گلبولهای قرمز میشود.
سرویسهای ارائهدهنده خدمات انتقال خون، نقش مهمی در تضمین پاسخدهی مناسب بیماران به تزریق
پلاکت بر عهده دارند. علاوه بر استفاده از پلاکتهای با HLA یکسان و سازگار به بیمارانی که دچار پاسخ آلو ایمنی گردیدهاند، حذف لکوسیت ها از محصولات خونی، سازگاری آنتیژنهای ABO و استفاده از پلاکتهای تازه(ذخیره شده زیر 48 ساعت)، باعث بهبود قابل مشاهدهای در میزان پلاکتها پس از انتقال میگردند. امروزه با بررسی فاکتورهای ایمنی مؤثر بر مقاومت پلاکتی، اطلاعات بسیاری جهت پیشگیری از بروز آن به دست آمده و اقدامات ضروری بیشتر از قبل جهت کنترل و مدیریت این بیماری انجام میپذیرد. با این وجود و علیرغم پیشرفتهای بسیار در مدیریت و پیشگیری از بروز پدیده آلو ایمنی ایجادکننده مقاومت پلاکتی، مدیریت فاکتورهای غیر ایمنی، شرایط نامطلوب بالینی و استفاده از داروهایی که بر روی بقا یا عملکرد پلاکتها تاثیرگذار میباشند، همچنان به عنوان مشکلات حل نشدهای در مسیر بروز مقاومت پلاکتی وجود دارند و کشورهای مختلف با توجه به امکانات پزشکی خود بایستی تمهیدات ویژهای جهت رفع این معضلات و مدیریت این بیماری ایجاد کنند.
متن کامل: (13217 مشاهده)
مقاومت پلاکتی و چگونگی ایجاد آن
سعیده میلانی1، فاطمه یاری2
چکیده
سابقه و هدف
تزریق پلاکت، یکی از درمانهای اصلی بیماران ترومبوسیتوپنیک، جهت کاهش شدت و فراوانی عواقب ناشی از خونریزی میباشد. عدم افزایش متناسب پلاکتها متعاقب تزریق آنها تحت عنوان مقاومت پلاکتی شناخته میشود. هدف از این مطالعه، بررسی مکانیسمهای دخیل در بروز و شکلگیری مقاومت پلاکتی بود.
مواد و روشها
این مقاله به مرور فاکتورهای مؤثر در افراد دریافتکننده و اهداکننده پلاکت و برخی از خصوصیات خود محصول پلاکتی در بروز مقاومت پلاکتی پرداخته است که از طریق بانکهای اطلاعاتی Science Direct ، PubMed ،Medline ،SID ،Scopus و Magiran و جستجوی کلید واژههای مقاومت پلاکتی، آلوآنتیبادی، HLA و تزریق پلاکت در بین حدود 130 مقاله مرتبط انجام شده و نهایتاً 97 مقاله برای نوشتن استفاده گردید.
یافتهها
مقاومـت پلاکتی به دو گروه مقاومت ایمونولوژیک و غیر ایمونولوژیک قابل تقسیم میباشد. نوع ایمونولوژیک عمدتاً با ایجاد آلوایمونیزاسیون ناشی از تماس بـا آنتـیژنهای لکوسیت انسانی -HLA و آنتیژنهای پلاکت انسانی HPA- ایجاد میگردد. در موارد غیر ایمونولوژیک که حدود80% علل بروز مقاومت پلاکتی به شمار مـیآیند، عواملی همچـون تب، بزرگی طحال و مصرف برخی داروها منجر به بروز مقاومت پلاکتی میگردند.
نتیجه گیری
شناسایی عوامل و مکانیسمهای دخیل در ایجاد مقاومت پلاکتی اعم از عوامل ایمونولوژیک و غیر ایمونولوژیک، نقش مهمی در پیشگیری، مدیریت بیماری و جلوگیری از گسترش و تکرار آن بر عهده خواهد داشت.
کلمات کلیدی: پلاکتها، تزریق خون، ترومبوسیتوپنی
تاریخ دریافت: 11/10/98
تاریخ پذیرش: 22/2 /99
1- مؤلف مسئول: PhD بیوتکنولوژی پزشکی ـ استادیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران ـ صندوق پستی: 1157-14665
2- PhD ایمونولوژی ـ استاد مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
سعیده میلانی1، فاطمه یاری2
چکیده
سابقه و هدف
تزریق پلاکت، یکی از درمانهای اصلی بیماران ترومبوسیتوپنیک، جهت کاهش شدت و فراوانی عواقب ناشی از خونریزی میباشد. عدم افزایش متناسب پلاکتها متعاقب تزریق آنها تحت عنوان مقاومت پلاکتی شناخته میشود. هدف از این مطالعه، بررسی مکانیسمهای دخیل در بروز و شکلگیری مقاومت پلاکتی بود.
مواد و روشها
این مقاله به مرور فاکتورهای مؤثر در افراد دریافتکننده و اهداکننده پلاکت و برخی از خصوصیات خود محصول پلاکتی در بروز مقاومت پلاکتی پرداخته است که از طریق بانکهای اطلاعاتی Science Direct ، PubMed ،Medline ،SID ،Scopus و Magiran و جستجوی کلید واژههای مقاومت پلاکتی، آلوآنتیبادی، HLA و تزریق پلاکت در بین حدود 130 مقاله مرتبط انجام شده و نهایتاً 97 مقاله برای نوشتن استفاده گردید.
یافتهها
مقاومـت پلاکتی به دو گروه مقاومت ایمونولوژیک و غیر ایمونولوژیک قابل تقسیم میباشد. نوع ایمونولوژیک عمدتاً با ایجاد آلوایمونیزاسیون ناشی از تماس بـا آنتـیژنهای لکوسیت انسانی -HLA و آنتیژنهای پلاکت انسانی HPA- ایجاد میگردد. در موارد غیر ایمونولوژیک که حدود80% علل بروز مقاومت پلاکتی به شمار مـیآیند، عواملی همچـون تب، بزرگی طحال و مصرف برخی داروها منجر به بروز مقاومت پلاکتی میگردند.
نتیجه گیری
شناسایی عوامل و مکانیسمهای دخیل در ایجاد مقاومت پلاکتی اعم از عوامل ایمونولوژیک و غیر ایمونولوژیک، نقش مهمی در پیشگیری، مدیریت بیماری و جلوگیری از گسترش و تکرار آن بر عهده خواهد داشت.
کلمات کلیدی: پلاکتها، تزریق خون، ترومبوسیتوپنی
تاریخ دریافت: 11/10/98
تاریخ پذیرش: 22/2 /99
1- مؤلف مسئول: PhD بیوتکنولوژی پزشکی ـ استادیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران ـ صندوق پستی: 1157-14665
2- PhD ایمونولوژی ـ استاد مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران
مقدمه
کلیاتی راجع به مقاومت پلاکتی:
ترومبوسیتوپنی به شرایطی گفته میشود که مقدار پلاکت(ترومبوسیت) های موجود در خون، پایینتر از حد طبیعی(150000 پلاکت در هر میکرولیتر خون) باشد(1). کم بودن پلاکت در بیماران دریافتکننده مداوم این فرآورده، تحت عنوان مقاومت پلاکتی شناخته میشود. شاخصهای رایج جهت محاسبه مقاومت پلاکتی شامل افزایش تعداد تصحیح شده CCI (Corrected Count Increment)، افزایش پلاکت پس از انتقال PPI (Post-transfusion Platelet Increment) و درصد بازیافت پلاکتی PPR (Percentage Platelet Recovery) میباشند(جدول 1). با این حال محاسبه CCI یکی از بهترین پارامترها جهت بررسی وجود مقاومت پلاکتی میباشد و mL /7500CCI< پس از حداقل دو تزریق پیاپی پلاکت حاوی آنتیژنهای ABO سازگار یا تقریباً یکسان، به عنوان مقاومت پلاکتی معرفی میگردد(2).
مطالعههای مختلف نشان میدهند که سیستم ایمنی خصوصاً ایمنی همورال، مسئول 20% موارد مقاومت پلاکتی میباشد و در این بین نقش آلوآنتیبادیهای تولیدی در حذف پلاکتها از اهمیت بسیاری برخوردار میباشد(7-3). در حقیقت این آنتیبادیها، با تخریب پلاکتهای حاوی آنتیژن ناسازگار، منجر به بروز مقاومت پلاکتی میگردند. در کنار عوامل ایمونولوژیک، عوامل و فاکتورهای غیر ایمونولوژیک نیز میتوانند منجر به بروز مقاوت پلاکتی گردند(8)(جدول 2). بر خلاف عوارض ناشـی از تخریـب گلبـولهای قرمـز، تخریب پلاکتها به
صورت طبیعی، نشانههای بالینی از خود نشان نمیدهد(9).
با آشکار شدن اهمیت و نقش آنتیبادیها در فرد دریافتکننده پلاکت بر ضد آنتیژنهای بیگانه فرد اهداکننده در بروز مقاومت پلاکتی، عمده توجهات روی آنتیبادیهای ضد(Human Leukocyte Antigen) HLA متمرکز گردیده است(2). در اوایل دهه 1990، شناسایی آنتیژنهای HLA افراد دهنده و گیرنده و سازگار بودن آنها در طی تزریق پلاکت مورد توجه قرار گرفت که این امر به بهبود مقاومت پلاکتی در زیر گروهی از بیماران منجر گردید. با این حال همچنان گروه بزرگی از بیماران در خطر بروز مقاومت پلاکتی میباشند. این امر خصوصاً در مورد بیماران دارای بدخیمیهای خونی از اهمیت خاصی برخوردار میباشد(10).
گزارشهای مربوط به بروز پاسخ آلوایمنی نسبت به آنتیژنهای HLA بسیار بیشتر از آنتیژنهای HPA (Human Platelet Antigen) بوده است و به نظر میرسد آلوآنتیبادی ضد HLA ، عامل اصلی بروز مقاومتهای پلاکتی باشد. سیستم HLA از کمپلکس عمده سازگاری بافتی(MHC) منشاء میگیرد که پروتئینهای سطحی پلیمورفیک مهم جهت عرضه آنتیژن را کد میکنند. در واقع MHC یک سیستم چند شکلی و چند ژنی میباشد و حاوی لوکوسهایی برای کد کردن آلوآنتیژنهای HLA کلاس Iو II است. این ژنها نزدیک به هم و بر روی بازوی کوتاه کروموزوم 6 قرار دارند. این کمپلکس شامل 4 میلیون جفت باز DNA میباشد و کدکننده آنتیژنهایHLA-A ، Bو C (کلاس I ) و HLA-DR ، DQ و DP (کلاس II ) میباشد(12، 11).
جدول 1: شاخصهای محاسبه مقاومت پلاکتی
جدول 2: دلایل بروز مقاومت پلاکتی
آلوآنتیبادیهای ضد HLA میتوانند به دنبال تزریق پلاکت، پیوند و در طی بارداری تشکیل گردند(15-13، 10). حذف لکوسیتها از محصولات پلاکتی به کاهش بیش از 50% آلوایمونیزاسیون ضد HLA منجر گردیده است، ولی با این حال در 20% تا 30% بیماران دریافتکننده پلاکت، همچنان آلوآنتیبادی تشکیل میگردد(17، 16). تیتر بالای آنتیبادیهای ضد HLA با مقاومت پلاکتی در این بیماران همراه میباشد(18). در 12% تا 15% این بیماران که نیازمند دریافت طولانی مدت پلاکت میباشند، مقاومت پلاکتی رخ داده و افزایش ناکافی در تعداد پلاکتها به علت حذف سریع پلاکتهای انتقالی اتفاق میافتد(18). جهت درمان بیماران دارای آلوآنتیبادی ضد HLA ، تزریق پلاکتهای سازگار با HLA انجام میپذیرد. با این حال پیدا کردن پلاکتهای سازگار با HLA در مقیاس زیاد برای این بیماران با مشکل همراه است. در حال حاضر تزریق پلاکت به بیماران دارای آلوآنتیبادی بر مبنای تزریق پلاکتهای فاقد آنتیژنهای مربوط به آلوآنتیبادیهای ضد HLA در بدن این افراد استوار است با این وجود بایستی به اثرات آلوآنتی بادیهای موجود در گردش خون نیز توجه کرد(19).
در مورد سازگاری آنتیژنهای ABO ، از آنجایی که این آنتیژنها به میزان بسیار کم بر روی پلاکتها بیان گردیده و کمتر از 2 میلیلیتر گلبول قرمز در یک واحد پلاکت تزریقی وجود دارد، سازگاری ABOدر بروز مقاومت پلاکتی از اهمیت چندانی برخوردار نمیباشد. با این حال در افراد نیازمند تزریق مداوم پلاکت، میزان در معرض قرار گرفتن با گلبولهای قرمز و آنتیژنهای ABO
پلاکتی ممکن است در حدی باشد که منجر به تولید آنتیبادی در فرد دریافتکننده و همولیز پلاکتی گردد. از طرف دیگر، هر چند بیان آنتیژنهای ABO بر روی پلاکت بسیار کم بوده و وجود سازگاری ABO در تزریق پلاکت زیاد قابل توجه نمیباشد، در بیمارانی که با وجود تزریق پلاکتهای با HLA سازگار، همچنان کارآیی مؤثری در افزایش تعداد پلاکت مشاهده نمیشود، بایستی به وجود این سازگاری توجه بیشتری گردد(22-20).
ریجکرز و همکارانش نشان دادند که زیر مجموعهای از آلوآنتیبادیهای ضد HLA میتوانند باعث فعال شدن پلاکتهای اهداکنندگان و فاگوسیتوز آنها توسط ماکروفاژها گردند که در این فرآیند FcγRIIa نقش مهمی بر عهده دارد(19). مشابه این فرآیند در ترومبوسیتوپنی پورپورا ((ITP و ترانسفوزیون مرتبط با آسیب حاد ریه (TRALI) دیده شده است(24، 23). مطالعهها همچنین نشاندهنده این حقیقت است که اثرات آلوآنتیبادی بیشتر بر مبنای شناسایی اپی توپ به جای میل اتصال(افینیتی) آن استوار میباشد. همچنین هندسه اتصال آنتیبادی میتواند روی نحوه فعال شدن کمپلمان اثرگذار باشد(25).
مواد و روشها
ایمنیشناسی بروز مقاومت پلاکتی:
پلاکتهای حاوی آنتیژنهای بیگانه میتوانند عمدتاً تحت دو مسیر عمده به بدن عرضه گردند. اولین مسیر در طی بارداری بوده که یا به وسیله خونریزیهای کوچک در زمانهای مختلف بارداری اتفاق افتاده و یا عمدتاً در طی گردش خون جفتی و دوره پری ناتال پدید میآید.
آنتیژنهـای سلولهـای جنیـن به بروز پاسخ ایمنی در
مادر منجر میگردد که باعث انتقال آنتیبادیهای مادری به جنین و تخریب پلاکتهای مرتبط با این آلوآنتیبادیها میشود. این امر منجر به بروز ترومبوسیتوپنی و خونریزی در جنین و نوزاد تازه متولد شده میشود. موارد شدیدتر آن منجر به خونریزی داخل مغزی و ناتوانی شدید متعاقب آن میگردد.
ترومبوسیتوپنی آلوایمیون نوزادی- جنینی(FNAIT ؛ Foetal neonatal thrombocytopenia alloimmune)، یکی از همین موارد میباشد. بیشترین گزارشهای مربوط به پاسخ ایمنی بدن بر ضد آنتیژنهای بیگانه و تولید آلوآنتیبادی در دوران بارداری، مربوط به آنتیژنهای HLA و عمدتاً کلاس Iآن میباشد (هر چند آنتیبادی ضد HLA کلاس II نیز میتواند در طی این دوره شناسایی گردد). بر اساس روشها و یافتههایی که جهت محاسبه میزان آنتیبادیها مورد استفاده قرار میگیرند، 17% از خانمها بعد از یک بارداری، آنتیبادی ضد HLA بیان میکنند و این میزان در بارداریهای بعدی نیز افزایش مییابد(26).
دومین دلیل بروز پاسخ ایمنی ضد پلاکتها، انتقال خون میباشد که برای اولین بار به وسیله روتچر در سال 1981 ارائه گردید(27). ایجاد ایمنی ضد آنتیژنهای پلاکتی بعد از انتقال خون کامل امکانپذیر است که البته این فرآیند در کشورهای توسعهیافته بسیار نادر است. یکی از مسائلی که بایستی به آن توجه بسیاری کرد، اهدای پلاکت تازه که از زمان ذخیره آن بیش از 48 ساعت نگذشته است میباشد. چرا که در طی ذخیره پلاکت، میزان برخی مارکرها هم چون P-selectin بر روی آن افزایش مییابد که میتواند منجر به واکنش پلاکت با لکوسیتهای فرد دریافتکننده پلاکت گردد(28). همچنین در طی فرآیند ذخیره پلاکت، به علت فعالیت متابولیک پلاکت و لکوسیتهای باقیمانده، مواد غذایی مصرف گردیده و محصولات متابولیکی مضر تولید میگردند. فاکتورهای انعقادی فعال شده، بقایای سلولی و آنزیمهای پروتئولیتیک موجود در پلاسما میتوانند بر روی پلاکت تاثیر منفی بگذارند(29). از طرف دیگر انتقال سلولهای قرمز خون به علت وجود لکوسیتها که میزان زیادی آنتیژنهای HLA کلاسI را بیان میکنند و این آنتیژنها بر روی پلاکت نیز موجود میباشند، منجر به ایجاد پاسخ ایمنی ضد آنتیژنهای HLA میگردند. این امر با حذف لکوسیتها از محصولات خونی قابل پیشگیری میباشد. پرتوتابی UV نیز باعث غیر فعال شدن لکوسیتها و کاهش ایمونیزاسیون میگردد. با این حال حتی حذف لکوسیتها(حضور در محصولات پلاکتی با مقادیر کمتر از 106) نیز همچنان میتوانند ایمنیزا باشند. به طور کلی در بیماران دریافتکننده پلاکت که به طور مکرر تزریقات پلاکتی دریافت میکنند و همین طور دریافتکنندگان مکرر سلولهای قرمز خون، نوعی پان- سیتوپنی مشاهده میگردد. این افراد اگر در خطر خونریزی شدید باشند، بایستی پلاسمای درمانی نیز دریافت کنند که محصولی خونی و مملو از ایمونوگلوبولینهای واکنشی میباشد. بیمارانی که در نتیجه تزریق پلاکت، سیستم ایمنی آنها تحریک گردد ضد آنتیژنهای پلاکتی، آنتیژنهای HLA و آنتیژنهای ABH ، آنتیبادی تولید میکنند. این افراد میتوانند ضد آنتیژنهای گلبولهای قرمز هم پاسخ ایمنی از خود نشان دهند. این امر خصوصاً برای برخی از آنتیژنهای گلبولهای قرمز همچون RhD که به شدت ایمونوژن میباشند از اهمیت بیشتری برخوردار است. همچنین برخی از بیماران میتوانند در طی پیوند بافت نیز پاسخ ایمنی بر ضد آنتیژنهای HLA کلاس Iاز خود نشان دهند(30). بروز پاسخ ایمنی ضد پلاکت دریافتی، به عوامل مختلفی همچون سلولهای انتقال یافته، زمینه ژنتیکی افراد دریافتکننده پلاکت، بیماریهای زمینهای و داروهای دریافتی در این افراد(خصوصاً داروهای مهارکننده سیستم ایمنی و داروهای مداخلهکننده) بستگی دارد. عوامل دیگر همچون محیط و شرایط میکروبی نیز در این امر میتوانند تاثیرگذار باشند(30).
یافتهها
با توجه به اهمیت آلوآنتیبادی ضد آنتیژنهایHLA در بروز مقاومت پلاکتی، مطالعههای بسیاری در زمینه دلایل حساس شدن افراد گیرنده پلاکت نسبت به آنتیژنهای HLA افراد اهداکننده انجام شده است که میتواند در شناسایی مکانیسمهای دخیل در حساس شدن سیستم ایمنـی افراد دریافتکننده پلاکت نسبت به این آنتیژنها بسیار کمککننده باشد.
عمدهترین علل و مکانیسمهای حساس شدن به HLA :
1- حساس شدن به HLA در طی بارداری:
همانگونه که پیشتر گفته شد، حساسیت به HLA نتیجه عرضه آلوآنتیژنهای بیگانه به سیستم ایمنی فرد میباشد. بارداری یکی از مهمترین دلایل حساس شدن به HLA است چرا که نیمی از ژنهای جنین، بیگانه(آلوژن) میباشد و این در حالی است که سیستم ایمنی مادر، جنین در حال رشد را به لحاظ ایمنی در فضای رحم تحمل میکند اما در مقابل آنتیژنهای پدری به عنوان مثال آنتیژنهای HLA ، پاسخ ایمنی همورال و سلولار ایجاد میکند. علت وجود چنین تناقضی در پاسخهای ایمنی مادر در دوران بارداری همچنان ناشناخته باقی مانده است(31). سلولهای سایتوتروفوبلاست و سینسیشوتروفوبلاست، مرزی بین سلولهای بافت مادر و جنین ایجاد میکنند. این سلولها فاقد HLA-A و HLA-B بوده ولی HLA-G که مولکول تیپیک HLA کلاس I می باشند را ترشح میکنند. این امر جنین را از لیز شدن به وسیله سلولهای کشنده طبیعی حفظ میکند(32). این مکانیسم همراه با چند مکانیسم سازگاری دیگر، جنین را از حمله به وسیله سیستم ایمنی مادر مصون نگه میدارد. با این حال در طی رشد جفت، سلولهای سین سیشوتروفوبلاست به تدریج دچار آپوپتوز گردیده، تجزیه میگردند و محتوای سلولی خود و از جمله DNA جنینی را به جریان خون مادر آزاد میکنند. DNA جنین 7 هفته پس از بارداری در پلاسمای مادر قابل ردیابی میباشد و غلظت آن جهت ژنوتایپینگ جنین کافی است(33). بقایای این سلولهای آزاد شده میتوانند باعث بروز پاسخ ایمنی ضد HLA کلاس I گردند. هر چند سلولهای دیگری هم در این مسیر هستند که میتوانند از رحم به جریان خون مادر وارد شوند.
امروزه شکلگیری آنتیبادی ضد آنتیژن RhD در مادران فاقد این آنتیژن که جنین آنها آنتیژن RhD را روی گلبولهای قرمز خود حمل میکند به اثبات رسیده است. تیتر بالای آنتیبادیهای مادری ضد آنتیژن RhD جنین میتواند منجر به بروز بیماری همولیتیک جنینی در جنین و همین طور نوزاد تازه متولد شده گردد. به همین ترتیب، لکوسیتهای جنینی میتوانند سیستم ایمنی مادر را جهت تولید آنتیبادی ضد آنتیژنهای جنینی مانند HLA تحریک کنند. بر خلاف آنتیبادیهای مادری ضد آنتیژنهای گلبولهای قرمز همچنین آنتیژنهای گلیکوپروتئینی پلاکت و آنتیژنهای گرانولوسیتها، آنتیبادیهای مادری ضد HLA جنینی، به جنین آسیب نمیرسانند(34). میزان حساس شدن به HLA در مادران باردار بستگی به دفعات بارداری دارد و با افزایش تعداد بارداریها این میزان افزایش مییابد. همچنین روشی که برای ردیابی آنتیبادیها به کار میرود نیز در تعیین میزان حساسیت تاثیرگذار است . فاصله زمانی بررسی آنتیبادیها در زنان نسبت به زمان آخرین بارداری آنها نیز مهم میباشد و در زنانی که مدت زمان زیادی از آخرین بارداری آنها گذشته باشد، تعداد آنتیبادیهای کمتری ضد HLA قابل ردیابی خواهد بود(35).
2- حساس شدن به HLA موجود در کنسانترههای پلاکتی:
محصولات خونی مقادیر زیادی از مولکولهای HLA را به دریافتکنندگان این محصولات منتقل میکنند. گلبولهای قرمز، پلاکتها و لکوسیتهایی که ممکن است همراه این محصولات منتقل گردند، حاوی مولکولهای HLA در غشای خود میباشند. پلاسما نیز حاوی مولکولهای HLA محلول میباشد. گلبولهای قرمز منبع اصلی HLA انتقالی نبوده و تنها حدود 90 مولکول HLA بر روی سطح هر گلبول قرمز(محدوده 550-40 (موجود میباشد(37، 36). در مقابل پلاکتها حامل تعداد زیادی مولکول HLA هستند(حدود 120000-50000 مولکول HLA به ازای هر پلاکت)، هر چند که سطح پلاکتها نسبت به سطح گلبولهای قرمز بسیار کوچکتر است(38). پلاکتها تنها مولکولهای کلاس I عمدتاً HLA-A و HLA-B را بیان میکنند(40، 39). برخی یافتهها حاکی از آن هستند که پلاکتها مولکولهای HLA رها شده به وسیله سایر سلولها به پلاسما را نیز جذب میکنند(41). در یکی از مطالعههای اخیر، HLA-A*0201 که پپتیدهای مختلفی مانند پپتیدهای مشتق شده از گلیکوپروتئین IX را در ناحیه شناسایی آنتیژن ارائه میکند، در پلاکتها شناسایی شده است. HLA-A*0201که به وسیله پلاکتها بیان میگردد، در مگاکاریوسیتها یا سلولهای پیشساز آنها سرهمبندی میشود(42).
3- حساس شدن به HLA موجود در پلاسما:
مولکولهای HLA به میزان زیادی در پلاسما یافت میگردند. غلظت پروتئینهای HLA کلاس I بین 1/4-25/0 میکروگرم در هر میلیلیتر پلاسما و با وزن مولکولی 55 کیلو دالتون تخمین زده شده است(44، 43). غلظت مولکولهای HLA کلاس II ، 11 میکروگرم در هر میلیلیتر پلاسما گزارش شده است(34). در نتیجه بین 1013×1/4- 1013× 25/0 مولکول HLA کلاس I ، بیش از1010× 6/9 مولکول آلبومین و بیش از1011× 09/2 مولکول HLA کلاس I/ ایمونوگلوبولین در هر میلیلیتر از پلاسمای تازه فریز شده(Fresh Frezen Plasma )، به بیماران منتقل میگردد(45). نقش HLA محلول در ایجاد آلوآنتیبادیها زیاد مورد مطالعه قرار نگرفته است. در یک مطالعه در بیماران دچار کم کاری کلیه که تنها پلاسما دریافت کرده بودند، HLA محلول منجر به تولید آنتیبادی گردید(46). مولکول HLA کلاس I محلول میتواند با مولکول LDL ترکیب گردد و پس از فاگوسیته شدن، تجزیه و به وسیله ماکروفاژها و سایر سلولهای فاگوسیتی عرضه شود(47). لنفوسیتهای B و T میتوانند با شناسایی پپتیدهای عرضه شده ضد آنها، پاسخ ایمنی ایجاد کنند. با این حال گزارشهایی در مورد خاصیت مهارکنندگی سیستم ایمنی توسط مولکولهای HLA محلول در موش ارائه شده است. در یک مطالعه، در موشهایی که تحت درمان با HLA-B7 نوترکیب محلول قرار گرفته بودند، سیستم ایمنی همورال ضد لکوسیتهای حامل HLA-B7 ، کاملاً متوقف گردید(47).
4- حساس شدن به HLA ناشی از لکوسیتها:
لکوسیتهایی که همراه گلبولهای قرمز و کنسانترههای
پلاکتی موجود هستند، عمدهترین علت حساس شدن به HLA به شمار میروند(49، 48). لنفوسیتهای خون محیطی حدود 250000 مولکول HLA کلاس I را بر سطح خود حمل میکنند(34). همانند پلاکتها و گلبولهای قرمز، لکوسیتها هم مولکولهای HLA کلاس II را حمل میکنند. لکوسیتها میتوانند مولکولهای HLA را به پلاسمای بیماران آزاد کنند که باعث برداشت و عرضه آنها به وسیله سلولهای عرضهکننده آنتیژن(APC) میگردد. مهمتر این که لکوسیتها قادر به حمل پروتئینهای co-stimulatory میباشند. لنفوسیتهای بیماران با شناسایی مولکولهای HLAآلوژن، به آنها متصل گردیده و سیستم ایمنی آنها فعال میگردد. مولکولهای co-stimulatory روی لکوسیت افراد اهداکننده، منجر به تحریک و فعال شدن لنفوسیتهای بیماران میگردد. در نتیجه استفاده از پلاکتها و سلولهای قرمز فاقد لکوسیت، یکی از عمدهترین اقدامات جهت کاهش و جلوگیری از حساس شدن به HLA در افراد دریافتکننده پلاکت و خون میباشد(51، 50).
مقاومت پلاکتی، فرآیندی پیچیده بوده و دارای ابعاد بسیاری میباشد. مطالعههای گسترده روی FNAIT نشان داده است که مادرانی با ژنوتیپ HLA خاص، مثل DRB4*01:01 و خصوصاً DRB3*01:01 با پپتیدهای مشتق از HPA1 بیشتر واکنش میدهند تا افرادی که فاقد این ژنوتیپ هستند. نتایج مشابهی در مورد پاسخ ایمنی ضد آنتیژنهای گلبولهای قرمز و آنتیژنهای عرضه شده ناشی از پیوند بافت به دست آمده است و ژنوتیپهای خاص HLA (و به صورت کمتر برخی از آنتیژنهای سازگاری MICA و MICB) با پاسخهای ایمنی قویتر به آنتیژنهای فرد اهداکننده همراه بودهاند(53، 52).
مکانیسم عمل آلوآنتیبادیهای ضد HLA در بروز مقاومت پلاکتی:
مطالعههای قبلی نشان دادهاند که زیر گروهی از آلوآنتیبادیهای ضد HLA و سرم بیماران دارای این آنتیبادی، باعث فعال شدن پلاکت وابسته به FcgRIIa و افزایش فاگوسیتوز آنها به وسیله ماکروفاژها میگردند. اما این که چه حجم از این فعال شدن منجر به حذف پلاکتها میشود، همچنان نامشخص باقی مانده است(54). فعال شدن پلاکتها به آزاد شدن آلفا گرانولها ، ظهور (CD62P) P-selectin در سطح پلاکت و شروع شکلگیری کمپلمان به وسیله C3b ، منجر میگردد. C3b مستقیماً به P-selectin موجود بر سطح پلاکت متصل شده و فعال شدن کمپلمان منجر به جایگیری آن روی پلاکت میگردد. در این شرایط مسیر آلترناتیو کمپلمان آغاز گردیده که به دنبال آن اتصال IgG و نشست متعاقب C1q صورت میگیرد. در مرحله بعد، اتصال C3b فعال شدن کمپلمان را تسهیل نموده که در نهایت منجر به شکلگیری کمپلکس MAC (membrane attack complex) که نام دیگر آن کمپلکس C5b-9 است میگردد(55). فعال شدن کمپلمان میتواند به فعال شدن آن در فاز مایع نیز منجر گردد که در آن کندروتین سولفات آزاد شده از پلاکتهای فعال شده هدف قرار میگیرند(56). از طرف دیگر اتصال C3 به پلاکتهای فعال باعث القای میانکنش بین لکوسیت و پلاکت میگردد(57). تجمعات IgG باعث تجمع پلاکتها میگردد که این فرآیند با اضافه شدن C1q تشدید میشود (58). اضافه کردن آنتیبادیهای مونوکلونال ضد بتا-2-میکروگلوبولین و Pan HLA در مقادیر زیاد، باعث اتصال C3b و سمیت وابسته به کمپلمان(CDC) در پلاکت میگردد(60، 59).
نقش آنتیژنهای پلاکتی(HPA) در بروز مقاومت پلاکتی:
همان طور که پیش تر نیز به آن اشاره شد، یکی از دلایل ایجاد مقاومت پلاکتی، تولید آلوآنتیبادی ضد آنتیژنهای پلاکتی میباشد. یکی از دلایل ایجاد آلوآنتیبادی ضد آنتیژنهای پلاکتی، وجود پلیمورفیسم در این آنتیژنها است. این پلیمورفیسمها عمدتاً به علت تغییرات تک آمینو اسیدی در گلیکوپروتئین موجود بر سطح پلاکتها، مانند اینتگرین GPIIB/IIIA و GPIA/IIA، GPIB/V/IX تحت عنوان رسپتور فاکتور ون ویلبرند، GPIV تحت عنوان رسپتور کلاژن و ترومبواسپوندین و گلیکوپروتئین GPI CD109 میباشد(61). تفاوت زیادی در پلیمورفیسمهای HPA در جمعیتهای مختلف وجود دارد و بیماران در طی بارداری یا تزریق پلاکت، در مقابل این آنتیژنها پاسخهای آلوایمنی ایجاد میکنند. شیوع آنتیبادیهای اختصاصی ضد پلاکتها بین 11%-2% بوده و این در حالی است که حذف یا کاهش لکوسیتها، روی میزان آنها بیتاثیر میباشد(62). هر چند این آنتیبادیها شیوع فراوانی نداشته و به لحاظ آماری باعث کاهش معناداری در CCI نمیگردد. بر خلاف ترومبوسیتوپنی آلوایمیون یا پورپورای پس از انتقال، که حضور آنتیبادی Anti-HPA-1a در آن غالب میباشد، آلوآنتیبادی تولیدی در بیماران دریافتکننده پلاکت، اختصاصاً آنتیژنهای HPA-5b و HPA-1b را شناسایی میکند(63). جالب این که 70% آنتیبادیهای اختصاصی پلاکت در دورهای از عفونت در فرد ظاهر شده و ناپایدار میباشند. همچنین 50% این آنتیبادیها مانند اتوآنتیبادیها عمل میکنند(64).
زیر لایههای تنظیمی ایجاد آلوآنتیبادی:
مکانیسمهای دقیق دخیل در ایجاد ایمنی در افراد مبتلا به مقاومت پلاکتی همچنان ناشناخته باقی مانده است چرا که تعداد زیادی از افراد دریافتکننده پلاکت و مادران با بارداریهای متعدد، پاسخهای ایمنی ایجاد نمیکنند. این امر میتواند دلایل مختلفی داشته باشد. پلاکتهای بیگانه (همچنین سلولهای قرمز و لکوسیتها) انواعی از آنتیژنهای کوچک و بزرگ را بر سطح خود ارائه میکنند که برخی به عنوان واریانتهای آنتیژنیک شناخته شده و برخی به عنوان الگوهای مولکولی مرتبط با پاتوژن PAMPs
به شمار میروند.
PAMPs به عنوان، نشانههای خطر بیولوژیکی غیر خودی باعث فعال شدن سلولهای فاگوسیتی، دندریتی و لنفوسیتهایB حساس شده میگردند. این امر باعث فعال شدن پاسخهای پیش التهابی ایمنی ذاتی شده که برای عرضه آنتیژن و فعال کردن لنفوسیتهای T ضروری است. پلاکتهایی که در هنگام دریافت سیگنالهای خطر در تماس با لکوسیتها فعال میشوند، مستعد ترشح مقادیر زیادی از مولکولهای پیش التهابی هستند که در مجموع تحت عنوان Biological Response Modifiers (BRMs) به شمار میروند(66، 65). برخی از این فاکتورها و فاکتورهای ناشناخته دیگر میتوانند با سلولهای دخیل در سیستم ایمنی ذاتی، عرضه آنتیژن و پاسخهای ایمنی اکتسابی تداخل نمایند. یکی از این BRM هایی که در مقادیر زیاد به وسیله پلاکتها تولید میگردد، CD40L میباشد که در حالت محلول sCD40L و متصل به غشاء میتوانند با سلولهای عرضهکننده آنتیژن دارای CD40 مانند لنفوسیتهای B متصل گردد(67). در مدلی آزمایشی، کشت همزمان پلاکتها و لنفوسیتهای B باعث فعال شدن آنها(افزایش بیان P-selectin روی پلاکت و CD86 روی لنفوسیت B) گردید. در حقیقت میانکنش لنفوسیتهای Bو پلاکتها با تغییر در بیان CD40 و CD40L به وسیله لنفوسیتهای B و پلاکتها شناخته میشود. سه روز پس از انکوباسیون لنفوسیتها با پلاکتها، لنفوسیتهای B تمایز یافته تولید IgG1 ،IgG2 و IgG3 (اما نه IgG4 ، IgM و IgA) را افزایش میدهند(68). همچنین پاسخ لنفوسیتهای B فراخوانی شده به دلیل فعال شدن همزمان به وسیله(PRRs ؛ Pathogen Recognition Receptors) و رسپتور سلول B برای آنتیژن تسهیل میشود. اتصال آنتیژن به وسیله آنتیبادی سبب تقویت لنفوسیتهای Bواکنشدهنده به وسیله رسپتورهای FC و رسپتورهای کمپلمان میگردد. از طرف دیگر ارائه آنتیژن جهت عرضه آن به لنفوسیتهای T واکنشدهنده مهم است تا این لنفوسیتها به عنوان عملگرهای کمکی، منجر به تداوم تمایز لنفوسیتهای B واکنشدهنده گردند. شناسایی آلوآنتیژنها میتواند به وسیله دو مکانیسم صورت پذیرد: نخست شناسایی مستقیم به وسیله رسپتورهای لنفوسیتهای T ضد پپتیدهای مشتق از HLA که به وسیله سلولهای عرضهکننده آنتیژن فرد اهداکننده صورت میگیرد که این فرآیند در هنگام حذف دقیق لکوسیتها و حذف APCهای بیگانه تا حدود زیادی قابل جلوگیری میباشد. دوم پپتیدهای محلول مشتق از HLA فرد اهداکننده که به APC های فرد گیرنده متصل میگردد. آزمایش بلوکه کردن تماس APC و لنفوسیتهای T واکنشدهنده با اتصال متقاطع CTLA4-Ig باعث مهار ایجاد پاسخ ایمنی میگردد(70، 69).
ساریـس و همکارانـش نشـان دادنـد کــه پلاکتهای
اندوسیتوز شده به وسیله سلولهای دندریتی، احتمالاً به وسیله مکانیسم آپوپتوز، منجر به تحریک تولید اینترفرون گاما به وسیله لنفوسیتهای T CD4+ میگردند. این مسأله نشان میدهد که نه تنها HLA مربوط به لکوسیتهای باقیمانده در محصولات خونی بلکه HLA کلاس I پلاکتهای بیگانه هم میتوانند ایمنیزا باشند(71).
مولکولهای MHC کلاس I پلاکتی:
در گردش خون، پلاکتها بیشترین سهم را از مولکولهای MHC کلاس I در گردش به خود اختصاص میدهند و مطالعهها نشان میدهند که حدود دو سوم این مولکولهای پلاکتی، به وسیله پلاسما جذب میشوند(72). این مولکولهای MHC جذب شده از برشهای پروتئولیتیک غشای سلولهای سوماتیک حاصل میگردند. به نظر میرسد بیشتر آنتیژنهای MHC موجود بر روی پلاکتها، عمدتاً حاوی زنجیرههای سنگین و بتا-2-میکروگلوبولین باشند(73). در نتیجه بیشتر مولکولهای MHC پلاکت از نظر ساختاری تغییر یافته و توانایی جدا شدن از غشای پلاکت به دنبال ذخیرهسازی پلاکت را دارا هستند. پیشنهاد میشود که چون مولکولهای محلول MHC به محض ذخیره افزایش مییابد و ممکن است این مولکولهای مشتق از پلاکت، مسئول واکنشهای ایمنی بعدی پس از تزریق پلاکت باشند. همچنین میتواند دلیل این باشد که چرا این مولکولهای MHC محرک ضعیف لنفوسیتهای T CD8+ هستند و باعث مهار این لنفوسیتها و افزایش مقاومت در پیوندهای پوستی آلوژن میگردند(42). با این وجود گرچه بیشتر MHC های پلاکتی تجزیه میگردند این مولکولها هنوز دارای توانایی بالقوه در تحریک تولید آلوآنتیبادی میباشند. با تزریق پلاکتهای آلوژن به گیرنده، میزبان در معرض مقادیر زیادی مولکولهای تغییر یافته MHC کلاس I فرد اهداکننده قرار میگیرد. مولکولهای MHC فرد اهداکننده، سرانجام در طی مسیر گردش در طحال فاگوسیته میشوند (مثلاً کلاس I مرتبط با پلاکت) یا از طریق پینوسیتوز (کلاس I محلول) به وسیله سلولهای(مثلاً ماکروفاژها) سیستم رتیکولواندوتلیال گرفته میشوند. این مکانیسم جذب اولیه ماکروفاژهای گیرنده طحال و سلولهای دندریتی، اجازه میدهد تا به عنوان سلولهای عرضهکننده آنتیژن جهت تحریک سیستم ایمنی اکتسابی و در نهایت تولید آنتیبادی ضد آنتیژنهای فرد اهداکننده عمل کنند(73).
تولید آلوآنتیبادی ضد پلاکتهای فاقد لکوسیت:
جهت تولید آلوآنتیبادی ضد آنتیژنهای پلاکتهای انتقالی، شناسایی غیر مستقیم آلوآنتیژنها در طحال نقش کلیدی بر عهده دارد(75، 74). APC های گیرنده در طحال ابتدا بایستی پلاکتهای آلوژن را جذب کرده و آنها را به نواحی اندوزومی پردازش آنتیژن، جهت برش پروتئولیتیک MHC های پلاکتی به پپتیدهای 10-5 آمینواسیدی منتقل کنند. پپتیدها سپس به شکاف MHC II متصل میشوند و از آنجا به سطح APC جهت ارائه لنفوسیتهای CD4+ T منتقل میگردند(76). همان طور که سلولهای T از طحال عبور میکنند، گیرندههای اختصاصی پپتید-MHC آنها، آنتیژنهای موجود بر سطح APC را بررسی کرده و اگر رسپتور لنفوسیت T تمایل کافی برای اتصال بهMHC - پپتید پلاکتی(سیگنال اول) داشته باشد و شرایط CO-Stimulatory (سیگنال دوم) هم موجود باشد، لنفوسیتهای T فعال شده و به سلول افکتوری تمایز مییابند(76). سایتوکاینهای ترشح شده از سلولهای T کمکی فعال شده، با تحریک لنفوسیتهای B هدفگیری شده ضد MHC کلاس I افراد دهنده، باعث تمایز آنها به پلاسما سلها میگردد که این امر به ترشح آنتیبادی IgG ، اتصال بعدی این آنتیبادیها به پلاکت فرد اهداکننده و تخریب آنها منجر میگردد. مطالعههای حیوانی نشان دادهاند که تزریق پلاکتهای آلوژن، حالت پیش التهابی را موجب گردیده و سایتوکاینهای مربوط به لنفوسیتهای T خصوصاً لنفوسیتهای T کمکی Th1 باعث تولید اینترفرون گاما میگردد(77).
بروز مقاومت پلاکتی ناشی از عوامل غیر ایمونولوژیک:
بـرخی شرایـط کلینیکـی مـیتواند نیـاز به پلاکتها را
افزایش داده یا به تخریب آنها منجر گردد. بر اساس مطالعههایی بر روی بیماران مبتلا به لوسمی میلوئیدی حاد یا بیمارانی که تحت پیوند سلولهای بنیادی خونساز قرار گرفتهاند، شرایطی همچون تب، سپسیس، بیماری پیوند در مقابل میزبان(GVHD)، انعقاد داخل عروقی منتشر، بزرگ شدن طحال و مصرف برخی داروها میتوانند منجر به مقاومت پلاکتی گردند. تزریق پلاکت در این بیماران ممکن است با افزایش کافی تعداد پلاکت همراه نباشد. مقاومت زمانی آشکار میگردد که حداقل پس از 2 انتقال پیاپی پلاکتهای تازه(به محض تولید یا نهایتاً 2 الی 3 روز پس از تولید) با آنتیژنهای ABO سازگار، تعداد پلاکتها در بیمار به تعداد قابل قبول نباشد. توافقی بر سر این که تعداد پلاکتها بایستی 1 ساعت پس از انتقال یا 24-20 ساعت پس از انتقال محاسبه گردد وجود ندارد. همچنین فرمولی برای تصحیح حجم خون بیماران و تعداد دفعات تزریق پلاکت در این شرایط وجود ندارد. با این حال یک راهکار کلینیکی ساده جهت شناسایی بروز مقاومت پلاکتی، بررسی افزایش کمتر از 109×10 پلاکت در یک لیتر خون در بازه زمانی 24-20 ساعت پس از تزریق پلاکت میباشد(78).
سپسیس:
سپسیس به عنوان پاسخ بدن به عوامل تهدیدکننده حیات آن گفته میشود که میتواند منجر به آسیب بافت، نارسایی در عملکرد اندامهای بدن و مرگ گردد. ارتباط بین سپسیس و ترومبوسیتوپنی به خوبی آشکار شده است هر چند علت این ارتباط به خوبی مشخص نمیباشد. هماتوفاگوسیتوز که در مغز استخوان بیماران دارای سپسیس و ترومبوسیتوپنی زیاد دیده میشود، میتواند یکی از فاکتورهای دخیل در بروز مقاومت پلاکتی ناشی از سپسیس باشد(79).
همچنین کاهش تولید پلاکت نیز در برخی بیماران مبتلا
به سپسیس میتواند دلیل بروز ترومبوسیتوپنی باشد. در نهایت میتوانند در سطح اندوتلیوم فعال شده متوقف گردند که نقش مهمی در پاسخ میزبان به سپسیس بر عهده دارد. علاوه بر هموستازی، پلاکتها نقش مهمی در ایمنی ذاتی و اکتسابی داشته و پلاکتها میتوانند با لکوسیتها و سلولهای اندوتلیومی میانکنش داشته و این میانکنش باعث سوق داده شدن لکوسیتها به سمت بافت ملتهب میگردد. میانکنش اولیه پلاکت و لکوسیتها با اتصال P-selectin روی سطح پلاکت با PSGL-1 روی لکوسیت شکل میگیرد. مهار فعل و انفعالات بین پلاکت ـ نوتروفیل میتواند آسیب ریوی حاد ناشی از سپسیس را نیز بهبود بخشد(80).
پلاکتها با بیان Toll-like receptor ها که ساختارهای مولکولی روی سطح عامل پاتوژن را شناسایی میکنند، باعث ایجاد پاسخهای پیش التهابی میگردند. این فرآیند میتواند منجر به ترومبوسیتوپنی نیز بشود(81). علاوه بر این، محصولات باکتریایی مانند لیپوپلی ساکاریدها میتوانند در تعامل با آنتیبادیهای پلاکتی باعث افزایش فاگوسیتوز گردند(82). این یافتهها نشان میدهند که چگونه فاکتورهای کلینیکی مانند سپسیس میتواند منجر به مقاومت پلاکتی در افراد دریافتکننده پلاکتهای دارای آلوآنتیژن گردد.
بزرگ شدن طحال:
طحال مهمترین عامل مؤثر بر CCI پلاکتی در فرآیند انتقال میباشد. در واقع طحال به عنوان یکی از مکانهای به دام انداختن پلاکت محسوب شده و طحالهای بزرگتر حاوی ذخایر پلاکتی بیشتری هم هستند. پلاکت بزرگ باعث کاهش فواصل زمانی تزریق پلاکت میشود. مطالعهها نشان دادهاند که طحال جایگاه عمده تخریب پلاکتی میباشد. بیش از 85% پلاکتها در طحال بیماران مبتلا به اسپلنومگالی تخریب میگردند اما این میزان در گروه کنترل طبیعی 61% میباشد.
در بیماران فاقد طحال، کبد محل اصلی تخریب پلاکت محسوب میگردد که 89% تخریب در آنجا صورت میگیرد. میزان بازیافت پلاکت در گردش اندکی پس از تزریق، 26% در بیماران با طحال بزرگ، 59% در گروه کنترل و 8/97% در بیماران فاقد طحال گزارش شده است. 30 دقیقه پس از انتقال، 80% پلاکتها در طحال بیماران مبتلا بـه طحـال بـزرگ مشاهـده گردیـده اسـت در حالـی که ایـن میـزان در طحـال افـراد طبیعـی حـدود 40% بـوده
است(83).
تب:
در بیماران مبتلا به بدخیمیهای خونی، تب عامل عمده ایجاد مقاومت پلاکتی میباشند. اگر چه بروز تب با کاهش CCI همراه است. معلوم نیست حضور عوامل ایجاد کننده پیچیدگی مانند عفونت یا داروها در این امر دخیل میباشد یا خیر. بررسی بیمارانی که ضد HLA فرد اهداکننده پاسخ ایمنی ایجاد کرده بودند نشان داد که حضور تب در هنگام تزریق پلاکت به اهداکنندگان، باعث کاهش PPR (Percent Platelet Recovery ) میگردد. با این حال نکته حائز اهمیت این مطلب زمانی آشکار شد که با استفاده از پلاکتهای سازگار، تب باعث اثر مشخصی روی PPR نگردید(84).
انعقاد درون عروقی منتشر (Disseminated intravascular coagulation):
مصرف فاکتورهای انعقادی و تولید نامنظم و بیش از حد ترومبین، باعث رسوب فیبرین در عروق کوچک میشود که به این فرآیند انعقاد درون عروقی منتشر یا DIC (Disseminated Intravascular Coagulation) گویند(85). بیماران مبتلا به لوسمی پرومیلوسیتیک حاد به علت رها شدن فاکتورهای بافتی از گرانولهای سلولهای لوسمی که یا به صورت خود به خودی یا در نتیجه شیمی درمانی اتفاق میافتد، DIC از خود نشان میدهند(85).DIC میتواند منجر به غیر فعال شدن و بیحرکت گردیدن پلاکتها شده و یکی از عوامل CCI ضعیف پس از تزریق پلاکت به شمار میرود(86).
سن پلاکت:
مطالعهها نشان میدهند سن پلاکت به طرز معناداری بر
روی CCI اثر گذار است. جالب این جـا اسـت که در یک مطالعه انجام شده بر روی بیماران دارای عفونت، انعقاد درون عروقی منتشر و بزرگی طحال، مقاومت نسبت به تزریق پلاکتهای کهنه در این بیماران ایجاد گردید در حالی که این فرآیند به هنگام تزریق پلاکتهای تازه در این افراد مشاهده نشد. یکی از دلایل این امر میتواند فعالشدن پلاکتهای قدیمی در طی چرخه ذخیرهسازی باشد. در نتیجه بایستی به سن پلاکتها به عنوان یک فاکتور غیر ایمونولوژیک در بروز مقاومت پلاکتی توجه کرد(87).
داروها:
ترومبوسیتوپنی ناشی از داروها شایع میباشد و داروهای مختلفی در این رابطه شناسایی گردیدهاند که از این میان میتوان به ونکومایسین، آمفوتریسین ب و هپارین اشاره کرد(88). داروها با مکانیسم ایمنی منجر به ترومبوسیتوپنی میگردند(89).
بیماری Venoocclusive و پیوند در مقابل میزبان، Graft Versus Host Disease (GVHD ):
بحثهای فراوانی راجع به این که پیوند سلولهای بنیادی خونساز عامل خطری برای مقاومت پلاکتی محسوب میگردد یا خیر وجود دارد. انسداد عروق کوچک کبدی(VOD: Veno Occlusive Disease) کبدی در 22% بیمارانی که پیوند سلولهای بنیادی خونساز دریافت کردهاند گزارش شده است. مقاومت پلاکتی میتواند یکی از علائم وخامت کلینیکی حال بیمارانی باشد که پیوند سلولهای بنیادی خونساز دریافت کردهاند(90). GVHD نیز فاکتور خطری برای بروز مقاومت پلاکتی در بیمارانی است که پیوند سلولهای بنیادی خونساز دریافت کردهاند. میزان بروز اتوآنتیبادیهای پلاکتی در بیماران با GVHD حاد یا مزمن به طرز معناداری بالاتر از گروه کنترل میباشد(92، 91).
التهاب و مقاومت پلاکتی:
پلاکتهـا قادر به تولید مدیاتورهای التهابی مثل IL-1a،
IL-8 ، RANTES ،TGFβ و همین طور CD154 هستند(93). CD154 های مشتق از پلاکت مستقیما قادر به تحریک لنفوسیتهای B جهت تکثیر و تولید آنتیبادی میباشند. در نتیجه حتی در صورت عدم حضور لنفوسیتهای T CD4+، پلاکتها میتوانند در القای تولید آلوآنتیبادی نقش کمکی داشته باشند(94).
برخـی مطالعههـا نشـان دادهاند که شرایط التهابی بیمار، زیر گروههای APC مصرفکننده گلبولهای قرمز را تغییر داده و منجر به بروز پاسخ ایمنی و تولید آلوآنتیبادی میگردند. اثر مشابهی ممکن است پس از تزریق پلاکت اتفاق بیفتد. بیماران دریافتکننده پلاکت معمولاً دارای آسیبهای قابل توجه همچون تروما، عفونت یا بیماری میباشند. مطالعه بر روی موشهایی که به لحاظ ژنتیکی یکسان بودند نشان داد که تفاوتهای محیطی هر حیوان مانند زمینه التهابی آن میتواند روی پاسخ ایمنی آنها مؤثر باشد(96، 95).
سایر عوامل:
برخی از مطالعهها حاکی از پایین بودن مقدار پلاکت تزریقی، پایین بودن کیفیت پلاکتهای تزریق شده و اندازه (size) بدن بیمار دریافتکننده پلاکت در بروز پاسخ ایمنی میباشند(97).
نتیجهگیری
علیرغم اقدامات جدی، مقاومت پلاکتی همچنان یک عارضه مهم در بحث انتقال خون و فرآوردههای خونی بوده که هر چند در گروه کوچکی از افراد مشاهده میگردد، اما پیشگیری از این عارضه در بیمارانی که نیازمند دریافت مداوم پلاکت میباشند همچنان امری مهم و حیاتی به نظر میرسد. در این راستا شناسایی عوامل ایجاد کننده مقاومت پلاکتی در جلوگیری از بروز آن از اهمیت به سزایی برخوردار میباشد. همان گونه که گفته شد، 80% موارد مقاومت پلاکتی ناشی از عوامل غیر ایمنی و 20% آن ناشی از عوامل ایمنی و در نتیجه تولید آنتیبادی ضد آنتیژنهای موجود بر سطح پلاکت میباشد. این آنتیبادیها میتوانند آنتیبادیهای خنثیکننده تولید شده ضد آنتیژنهای ABO موجود بر روی پلاکت، یا آلوآنتیبادیهایی باشند که ضد آنتیژنهای لکوسیت انسانی(HLA) کلاس I در 80% موارد، یا آنتیژنهای پلاکت انسانی(HPA) در 20% موارد بوده و منجر به تخریب پلاکتهای انتقالی گردند. یکی دیگر از راههای تولید آلوآنتیبادی ضد آنتیژنهای HLA ، پیوند بافت میباشد. در بارداری نیز، آنتیبادی ضد آنتیژنهای لکوسیت انسانی(HLA) که بر سطح سلولهای جنین آنها(به ارث رسیده از پدر) تولید میگردد میتواند در بارداریهای بعدی یا تزریق پلاکت در خانمهای دارای سابقه بارداری ایجاد مشکل کند. پلاکتها هم چنین آنتیژنهای پلاکتی(HPA) را بیان کرده که واریانتهایی از مولکولهای تجمع یافته و چسبنده میباشند و بین مادر و جنین و فرد اهداکننده و دریافتکننده متفاوت بوده که منجر به تولید آلوآنتیبادیها و مشکلات بعدی آن میگردد. تزریق پلاکت با انتقال لکوسیتها نیز همراه است که از طریق بیان کپیهای فراوان HLA کلاس I (و کلاس II بیان شده بر روی لنفوسیتهای T فعال شده، لنفوسیتهای B و سلولهای دندریتی) به تولید پاسخ ایمنی منجر میگردند. همچنین سلولهای قرمز خونی باقیمانده در کنسانترههای پلاکتی نیز باعث تولید آلوآنتیبادی ضد آنتیژنهای گلبولهای قرمز میشود.
سرویسهای ارائهدهنده خدمات انتقال خون، نقش مهمی در تضمین پاسخدهی مناسب بیماران به تزریق
پلاکت بر عهده دارند. علاوه بر استفاده از پلاکتهای با HLA یکسان و سازگار به بیمارانی که دچار پاسخ آلو ایمنی گردیدهاند، حذف لکوسیت ها از محصولات خونی، سازگاری آنتیژنهای ABO و استفاده از پلاکتهای تازه(ذخیره شده زیر 48 ساعت)، باعث بهبود قابل مشاهدهای در میزان پلاکتها پس از انتقال میگردند. امروزه با بررسی فاکتورهای ایمنی مؤثر بر مقاومت پلاکتی، اطلاعات بسیاری جهت پیشگیری از بروز آن به دست آمده و اقدامات ضروری بیشتر از قبل جهت کنترل و مدیریت این بیماری انجام میپذیرد. با این وجود و علیرغم پیشرفتهای بسیار در مدیریت و پیشگیری از بروز پدیده آلو ایمنی ایجادکننده مقاومت پلاکتی، مدیریت فاکتورهای غیر ایمنی، شرایط نامطلوب بالینی و استفاده از داروهایی که بر روی بقا یا عملکرد پلاکتها تاثیرگذار میباشند، همچنان به عنوان مشکلات حل نشدهای در مسیر بروز مقاومت پلاکتی وجود دارند و کشورهای مختلف با توجه به امکانات پزشکی خود بایستی تمهیدات ویژهای جهت رفع این معضلات و مدیریت این بیماری ایجاد کنند.
نوع مطالعه: مروري |
موضوع مقاله:
انتقال خون
ارسال پیام به نویسنده مسئول
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |
