تولید رده سلولی K562 بیان کننده اندونوکلئاز Cas9 (CRISPR-associated9)
فائزه انصاری1، مهین نیکوگفتار ظریف2، امیر علی حمیدیه3، مهدی شمسآرا4
چکیده سابقه و هدف امروزه ژنوم ردههای سلولی به منظور ایجاد مدلهای بیماری و درمان آنها ویرایش میشود. البته بزرگی ژن Cas9 موجب مشکلاتی از جمله پایین بودن کارآیی سیستم CRISPR شده است. برای حل این مشکل، ردههای سلولی بیانکننده Cas9 تولید شدهاند که در آنها تنها باید بخش RNA راهنمای CRISPR به سلول ترانسفکت شود. مواد و روشها در این مطالعه تجربی، قطعه PGK-PURO/CMV (PPC) با PCR از وکتور pAAVS1-puro-DNR تکثیر شده و در وکتور pTG19-Tهمسانهسازی شد. سپس قطعه PPC از این وکتور با آنزیمهای KpnI و EcoRI خارج شد. وکتور pCas-Guide-AAVS1 نیز تحت برش آنزیمی مشابه قرار گرفت و اتصال آن با قطعه PPC منجر به ساخت وکتور pPPC-Cas گردید. پس از بهینه کردن شرایط الکتروپوریشن، وکتور pPPC-Cas به درون سلولهای K562 الکتروپوریت شد و سلولهای مقاوم به پرومایسین انتخاب شدند و میزان بیان Cas9 در آنها با Real-time PCR بررسی شد. یافتهها با PCR قطعهای به طول 2514 جفت باز تکثیر شد. وکتورpPPC-Cas طی دو مرحله همسانهسازی ساخته شد. سلولهای ترانسفکت شده مقاوم به پرومایسین انتخاب شدند. انتخاب کلونی در ادامه انجام شد و سه کلونی که نسبت به هم بیانهای بالا، متوسط و پایین داشتند، انتخاب شدند. نتیجه گیری در این مطالعه سلولهای K562 بیانکننده Cas9 به دست آمدند که از آنها میتوان در مطالعههای آتیژنومیک عملکردی و نیز تولید مدلهای سلولی بیماریهای انسانی استفاده کرد. کلمات کلیدی:رده سلولی، الکتروپوریشن، ویرایش ژنی
تاریخ دریافت: 12/6 /97 تاریخ پذیرش: 25/10/97
1- کارشناس ارشد زیست فن آوری پزشکی ـ مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران 2- دکترای خونشناسی و بانک خون ـ دانشیار مرکز تحقیقات انتقال خون ـ مؤسسه عالی آموزشی و پژوهشی طب انتقال خون ـ تهران ـ ایران 3- فوق تخصص هماتولوژی، انکولوژی و پیوند سلولهای بنیادی ـ مرکز طبی کودکان ـ دانشگاه علوم پزشکی تهران ـ تهران ـ ایران 4- مؤلف مسئول: دکترای ژنتیک ـ استادیار پژوهشکده زیست فناوری کشاورزی ـ پژوهشگاه ملی مهندسی ژنتیک و زیست فناوری ـ همت غرب ـ تهران ـ ایران ـ صندوق پستی: 161/14965
مقدمه امروزه سیستمهای ویرایش ژن برای مقاصدی چون شناسایی نقش ژنها، تولید موجودات مدل، تولید ردههای سلولی دستکاری شده، واکسن و درمان بیماریها بسیار مورد توجه میباشند(5-1). در این میان سیستم CRISPR/Cas به علت سادگی، ارزانتر بودن و محدودیتهای کمتر در استفاده، گوی سبقت را از فناوریهای رقیب نظیر ZFN (Zinc finger nuclease)و TALEN (Transcription activator-like effector) ربوده است(6). برای نمونه، با ظهور روشهای ویرایش ژنوم، گزارشهایی مبنی بر ویرایش ژن بتا-گلوبین و یا فعال نمودن هموگلوبین جنینی با این روشها وجود دارد(7). سیستم CRISPR در واقع بخشی از سیستم ایمنی اکتسابی پروکاریوتها است. انواع مختلفی از سیستمهای CRISPR در باکتریها شناسایی شدهاند. اما پرکاربردترین سیستم به منظور ویرایش ژنوم، سیستم CRISPR/Cas9 است که از دو جزء آندونوکلئاز Cas9 و RNA راهنما (sgRNA) تشکیل شده است. به منظور ویرایش ژنی، ابزار CRISPR به اشکال مختلف در قالب پلاسمید، RNA و کمپلکس ریبونوکلئوپروتئین قابل استفاده است. پلاسمیدهای بیانکننده نوکلئاز Cas9و RNA راهنما، سادهترین و ارزانترین شکل استفاده از CRISPR هستند که در آن پلاسمید به تنهایی به داخل سلول هدف وارد میشود. اما به دلیل بزرگبودن ژن رمزکننده Cas9 (حدود 4 کیلو جفت باز) کارآیی ترانسفکشن این دست پلاسمیدها معمولاً پایین است. برای حل این مشکل امروزه ردههای سلولی بیانکننده Cas9 را توسعه دادهاند. مزیت استفاده از این ردههای سلولی آن است که لازم نیست به همراه sgRNA ، ژن Cas9 هم وارد سلول شود و بدین ترتیب قابلیت و کارآیی سیستم CRISPR/Cas در ویرایش ژنوم افزایش مییابد. لذا این دست سلولها، تبدیل به ابزار مفیدی در شناسایی اهداف دارویی و درمانی و نیز مطالعه عملکرد ژنها شدهاند. با توجه به تمام مزیتهایی که فناوری CRISPR/Cas9 دارد، در این مطالعه سعی شده است تا به منظور اثر بخشی بهتر آن، یک رده سلولی K562 بیانکننده ژنCas9تولید شود تا در مطالعههای آینده مرتبط مورد استفاده قرار بگیرد. رده سلولی K562 یک رده سلولی است که از بیماران لوسمی میلوئیدی مزمن(CML) گرفته میشود و برای مطالعههای مربوط به بیماریهای خونی یک رده سلولی مناسب میباشد(8). نشان داده شده است که استفاده از این ردههای سلولی موجب میشود تا کل فرآیند اصلاح ژنی سریعتر با صرف هزینه کمتر و با تاثیرگذاری بیشتر صورت بگیرد(9). با توجه به همه موارد مطرح شده، هدف از مطالعه حاضر تولید رده سلولی K562 بیانکننده اندونوکلئاز Cas9 (CRISPR-associated9) با هدف انجام مطالعههای مرتبط با بیماریهای خونی بود.
مواد و روشها این مطالعه تجربی بوده و بر روی رده سلولی K562 انجام گردیده است و دارای دو بخش مولکولی و سلولی میباشد.
بخش مولکولی کشت باکتری: برای کشت باکتری در محیط (Luria- Bertani) LB ، مایع حاوی سدیم کلراید، تریپتون، عصاره مخمر، آب، ساخته شده در محیط آزمایشگاه تحت شرایط استریل یک کلنی از باکتری E.coli در لوله آزمایش دارای 5 میلیلیتر محیطLB حاوی آنتیبیوتیک مناسب تلقیح شد. سپس فالکون به مدت 17 ساعت در انکوباتور 37 درجه سانتیگراد در rpm 150 شیک گردید.
استخراج پلاسمید در مقیاس کوچک(Mini-prep): DNA پلاسمید به روش لیز قلیایی(Mini-Prep Plasmid DNA extraction) با استفاده از کیت استخراج پلاسمید و بر اساس دستورالعمل شرکت یکتا تجهیز آزما، استخراج گردید.
وکتورهای استفاده شده: در ایـن مطالعـه وکتـورهـای pAAVS1-Puro-DNR به
جدول 1: توالی نوکلئوتیدی آغازگرهای استفاده شده در این مطالعه
سکانس هدف
جلوبرنده(5’-3’)
معکوس(5’-3’)
سایز آمپلیکون
PPC
CCGGAATTCCAGCTAGTCTTCTTCCTCCAAC*
GCAGATCTCCTCGGTACCGGATCCAGTCGACAAATTC*
bp 2514
B2M
CGCTACTCTCTCTTTCTGG
GTCAACTTCAATGTCGGATGGAT
bp 143
Cas9 trans
CCCCAAGAAAAAACGCAAGGTG
GTTCAGGAAACAGCTATGACCG
bp 181
*خط زیر نشاندهنده جایگاه برش آنزیمی EcoRI در آغازگر بالادست(جلوبرنده) و KpnI در آغازگر پاییندست(معکوس) است.
عنوان DNA الگو برای تکثیر قطعه PGK-Puro-CMV (پروموتر ژن فسفوگلیسرات کیناز، ژن مقاومت به پرومایسین و پروموتر ویروس سیتومگالو)، وکتور pTG19-T برای همسانهسازی محصول PCR قطعه PGK-Puro-CMV، وکتور pCas-Guide-AAVS1 برای زیر همسانهسازی(Subcloning) قطعه PGK-Puro-CMV و وکتور pEGFP-C1 به منظور بهینهکردن شرایط الکتروپوریشن مورد استفاده قرار گرفت.
طراحی آغازگر: طراحی آغازگر برای تکثیر قطعه PPC از پلاسمید هدف و نیز سنجش کمی بیان ژن Cas9 منتقل شده به سلولهای K562 استفاده شد. آغازگرها با استفاده از نرمافزار OLIGO7 طراحی شدند و از لحاظ ساختارهای ثانویه، تشکیل دایمر و نقطه ذوب(Tm) بررسی گردیدند. اختصاصی بودن آغازگرها نیز در سایت NCBI (Primer-BLAST) مورد ارزیابی قرار گرفت(جدول 1).
واکنش زنجیرهای پلیمراز(PCR): واکنـش زنجیرهای پلیمراز به منظور تکثیر قطعه PGK-Puro-CMV (به اختصار PPC)از وکتور pAAVS1-puro-DNR با استفاده از کیت Taq DNA Pol. 2X Master Mix از شرکت آمپلیکون طبق برنامه زمانی دمایی(یک چرخه دناتوره شدن اولیه 3 دقیقه و 30 ثانیه در 95 درجه سانتیگراد، 30 چرخه دناتوره شدن ثانویه 20 ثانیه در 95 درجه سانتیگراد، اتصال 30 ثانیه در58 درجه سانتی گراد، پلیمریزه شدن رشته DNA دو دقیقه و 30 ثانیه در 72 درجه سانتیگراد و یک چرخه پلیمریزه شدن نهایی 15 دقیقه در 72 درجه سانتیگراد) در ترمال سایکلر انجام شد و بـه منظـور تخلیص محصول واکنش زنجیرهای پلیمراز، از کیت استخراج از ژلGel and PCR Product Purification Mini Kit ساخت شرکت یکتا تجهیز آزما استفاده شد.
واکنش اتصال(Ligation): در این مطالعه واکنش اتصال یک بار به منظور همسانهسازی محصول PCR (قطعه PPC) در وکتور pTG19-T و بار دیگر برای زیر همسانهسازی قطعه PPC از وکتور pTG19-T به درون وکتور pCas-Guide-AAVS1 مورد استفاده قرار گرفت. در واکنش اتصال از آنزیمT4 DNA ligase استفاده شد و نسبت مولی vector:insert به صورت 1:3 در نظر گرفته شد. پس از آمادهسازی واکنش اتصال در یک لوله، به مدت 2 ساعت در دمای 22 درجه سانتیگراد و 14 ساعت در دمای 16 درجه سانتیگراد انکوبه گردید. محصول PCR در جایگاه کلونینگ پلاسمید pTG19-T همسانهسازی شد. محصول واکنش اتصال، به باکتری E.coli سویه DH5α ترانسفورم شد. به طور خلاصه، 10 میکرولیتر از واکنش اتصال به باکتری مستعد اضافه و به آرامی با هم مخلوط گردید و به مدت 30 دقیقه روی یخ گذاشته شد. به مخلوط فوق در دمای 42 درجه سانتیگراد به مدت 90 ثانیه شوک حرارتی داده شد و بلافاصله بر روی یخ منتقل گردید. برای تایید حضور قطعه مورد نظر در وکتور pTG19-T از کلونیهای به دست آمده، پلاسمید استخراج شد و با آنزیمهای KpnI و EcoRI برش داده شد. خروج قطعه PPC نشانه موفقیت در همسانهسازی ژن بود. هم چنین وکتور نوترکیب با استفاده از آغازگرهای عمومی M13 وT7 به روش سنگر توالییابی شد. نتایج تعیین توالی با نرمافزار Fich TV بررسی شدند. در ادامه به منظور زیر همسانهسازی قطعه PPC در وکتور بیانی، هضم آنزیمی EcoRI و KpnI روی پلاسمید حامل قطعه PPC و وکتور pCas-Guide-AAVS1 انجام شد. واکنش اتصال بین قطعات PPC و وکتور بیانی خطی انجام شده و پس از ترانسفورماسیون باکتری مستعد، پلاسمیدهای نوترکیب با هضم آنزیمی و خروج قطعه هدف شناسایی شدند.
استخراج پلاسمید در مقیاس زیاد(Maxi-Prep) : پس از تایید همسانهسازی ژن در وکتور پستاندار، برای الکتروپوریـت کردن وکتور pPPC-Cas به سلولهای K562 ، استخراج پلاسمید در مقیاس زیاد توسط کیتPlasmid DNA Extraction Maxi kit ساخت شرکت یکتا تجهیز آزما و مطابق با دستورالعمل آن انجام شد.
بخش سلولی: در این مطالعه رده سلولی K562 از مرکز ملی ذخایر ژنتیکی و زیستی ایران تهیه شد. محیط کشت مناسب استفاده شده برای رشد این سلولهای معلق(جیبکو) Medium 1640RPMI به همراه 10% FBS (سرم جنین گاوی) و 1% آنتیبیوتیک پنیسیلین/ استرپتومایسین بوده و طبق دستور ATCC شرایط محیطی کشت دمای 37 درجه سانتیگراد، تحت شرایط 5% CO2 و 95% رطوبت میباشد.
ترانسفکشن سلولهای K562 : یکی از روشهای انتقال وکتورهای پلاسمیدی یا یک DNA خارجی به درون سلول، الکتروپوریشن (Electroporation) میباشد. برای الکتروپوریشن تعداد 5 تا 6 میلیون سلول K562 استفاده شد. شمارش سلولها به کمک لام نئوبار(هموسایتومتر) انجام و برای بررسی درصد سلولهای زنده از رنگ تریپانبلو استفاده گردید. الکتروپوریشن با دستگاه بیوراد با ولتاژها و ظرفیتهای خازنی متفاوت انجام شد تا شرایط بهینه الکتروپوریشن مشخص شود. به منظور بهینه کردن شرایط الکتروپوریشن، از وکتور تجاری pEGFP-C1 ساخت شرکت کلونتک استفاده شد. سپس سلولها با وکتور ساخته شده در این مطالعه تحت شرایط بهینه شده الکتروپوریشن ترانسفکت شدند و سلولهای مقاوم به پرومایسین به مدت دو هفته در حضور آنتیبیوتیک انتخاب شدند. با توجه به تصادفی بودن ورود وکتور به داخل ژنوم سلول، به منظور دستیابی به یک جمعیت سلولی هموژن(همگن)، انتخاب کلونی (clonal selection) انجام شد.
استخراج RNA : RNA کل سلولهای K562 با استفاده از کیت ساخت شرکت یکتا تجهیز آزما، طبق دستورالعمل سازنده استخراج شد. برای تعیین غلظت و خلوص RNA استخراج شده، جذب نمونهها به وسیله نانو دراپ در طول موج 260 و 280 نانومتر خوانده شد. خلوص نمونهها از طریق نسبت جذب اسپکتروفتومتری 260:280 تعیین شد. همچنین تمامیت RNA استخراج شده بر روی ژل آگارز و مشاهده باندهای RNA ریبوزومی S 28 و S18 بررسی شد.
Real-time PCR : رشته اول cDNA با استفاده از کیت شرکت یکتا تجهیز آزما و آغازگرهای پایین دست ژنهای Cas9 و B2Mمطابق با دستورالعمل کیت انجام شد. در ادامه، برای بررسی کمی بیان ژن Cas9 در سلولهایی که به روش انتخاب کلونی جدا شده بودند، Real-time PCR با کیت شرکت یکتا تجهیز آزما و در دستگاه Mic qPCR ساخت شرکت بیو مولکولار سیستم انجام شد. برنامه زمانی دمایی دستگاه به صورت یک چرخه برای دناتوره شدن اولیه در دمای 94 درجه سانتیگراد و به مدت 4 دقیقه انجام گرفت. در ادامه، 40 چرخه برای دناتوره شدن با دمای 94 درجه سانتیگراد(25 ثانیه) و اتصال آغارگرها و پلیمریزه شدن رشته DNA در دمای 58 درجه سانتیگراد (30 ثانیه) تنظیم شد. واکنش در حجم نهایی 10 میکرولیتر شامل 5 میکرولیتر از Real-time PCR 2X Master mix، 2 پیکو مول از هر آغازگر، 1 میکرولیتر از cDNA الگو و آب مقطر تا حجم نهایی 10میکرولیتر آماده شد. ژن بتا 2- میکروگلوبولین (B2M) به عنوان ژن خانهدار مورد استفاده قرار گرفت. میزان بیان نسبی کلونها نسبت به هم با محاسبه چرخههای آستانه(Ct) مطابق فرمول DCt=Ctcasq-Ctb2mتعیین شد.
نحوه جمعآوری اطلاعات و تجزیه و تحلیل دادهها: تجزیه و تحلیل دادههای Real-time PCR با نرم افزار GraphPad و با استفاده از آزمون t-test بررسی شدند. سطح معنادار بودن نتایج به صورت 05/0 p≤ در نظر گرفته شد.
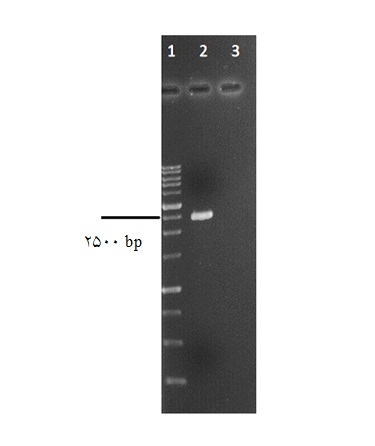
یافتهها بخش مولکولی: تکثیر قطعه PGK-Puro-CMV از وکتور pAAVS1-puro-DNR : قطعه PGK–Puro-CMV (PPC) با استفاده از آغازگرهای طراحی شده از وکتورpAAVS1-puro-DNR به وسیله PCR تکثیر شد. محصول PCR در کنار مارکر وزن مولکولی بر روی ژل آگارز 1% مورد بررسی قرار گرفت و قطعه تکثیر شده به طول 2514 جفت باز روی ژل آگارز مشاهده شد(شکل 1). همسانهسازی قطعه PPC در ناقلهای کلونینگ و بیانی: طی دو مرحله همسانهسازی، ابتدا محصول PCR قطعه PPC در وکتور کلونینگ و سپس در وکتور بیانی پستاندار وارد شد. همسانهسازی در وکتور کلونینگ pTG19-T بدون هضم آنزیمی و استفاده از وجود آدنین اضافه در انتهای محصول PCR ، به واسطه تکثیر با آنزیم Taq پلیمراز انجام شد. نتیجه همسانهسازی در وکتور کلونینگ، ابتدا با PCR از روی پلاسمید استخراج شده با آغازگرهای اختصاصی قطعه PPC تایید شد. نتیجه PCR از روی پلاسمید نوترکیب مشاهده باندی 2514 جفت باز معادل همان قطعه PPC روی ژل آگارز بود. در صورتی که در PCR از روی وکتور pTG19 به عنوان کنترل منفی، باندی مشاهده نشد(شکل 2). در ادامه وکتور نوترکیب با آنزیمهای تعبیه شده در طرفین قطعه KpnI و EcoRI برش خورده و خروج قطعه PPC به طول 2514 جفت باز از بدنه ناقل، به طول 2880 جفت باز، بر روی ژل آگارز مشاهده شد(شکل 3). در سطح مولکولی، عدم وجود جهش احتمالی در حین تکثیر قطعه PPC با روش توالییابی سنگر و مقایسه آن با توالی وکتور pAAVS1-puro-DNR بررسی شد. توالییابی با آغازگرهای عمومی M13 و T7 نشان داد که جهشی در قطعه تکثیر شده PPC رخ نداده است.
شکل 1: نتیجه تکثیر قطعه PPC به طول 2514 جفت باز به روشPCR . چاهک1: نشانگر وزن مولکولی 1 کیلو جفت بازی Thermo Scientific . چاهک2: قطعه تکثیر شده PPC
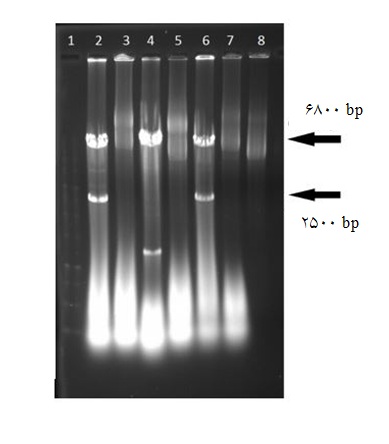
زیر همسانهسازی قطعه PPC در ناقل pCas-Guide-AAVS1 : در ادامـه، زیـر همسانـهسـازی قطعـه PPC از وکتــور کلونینگ به درون وکتور بیانی پستاندار انجام شد. وکتور pCas-Guide-AAVS1 با آنزیمهای EcoRI و KpnI برش خورد. در نتیجه این برش آنزیمی قطعهای به طول 1146 جفـت بـاز از وکتـور خـارج شـد. باند سنگینتر به طول
شکل 2: تایید همسانهسازی قطعهPPC در ناقل pTG19-T با روش هضم آنزیمی. چاهک1: نشانگر وزن مولکولی 1کیلو جفت بازی. چاهک 2: پلاسمید نوترکیب (pTG19-PPC) پس از برش با آنزیمهایKpnI و EcoRI .چاهک 3: پلاسمید برش نخورده وکتور pTG19-PPC
3: تایید همسانهسازی قطعه PPC در ناقل pTG19-T با روش PCR . چاهک 1: نشانگر وزن مولکولی 1 کیلو جفت بازی. چاهک 2: محصول PCR از روی پلاسمید سنگین تر (نوترکیب). چاهک 3: محصول PCR از روی پلاسمید سبک تر(پلاسمید غیرنوترکیب pTG19). تقریبی 8/6 کیلو جفت باز از روی ژل استخراج شد. وکتور کلونینگ حامل PPC نیز با این دو آنزیم برش داده شد. واکنش اتصال بین قطعات برش خورده PPC و ناقل گذاشته شده و بعد از ترانسفورماسیون، کلونیهای نوترکیب انتخاب شدند. موفقیتآمیز بودن کلونینگ ابتدا به روی کلونی PCR بررسی و مورد تایید قرار گرفت. تکثیر قطعه 2514 جفت بازی در برخی از کلونیها مشاهده شد(نتایج نشان داده نشده است). کلونهای دارای نتیجه مثبت و برخی از کلونهای منفی برای استخراج پلاسمید و هضم آنزیمی کشت شدند. هضم آنزیمی با KpnI و EcoRI منجر به خروج قطعه PPC در کلونهایی شد که نتیجه کلونی PCR آنها مثبت بود(شکل 4). پلاسمید نوترکیب حاصل به اختصار pPPC-Cas نامگذاری گردید.
شکل 5 : الف) درصد ترانسفکت شدن سلولهای K562 با شرایط مختلف الکتروپوریشن(Voltage-µF). اعداد نشاندهنده ولتاژ و ظرفیت خازنی مورد استفاده در روش الکتروپوریشن میباشند. بهترین شرایط ولتاژ 269 ولت و ظرفیت خازنی 1050 میکروفاراد است. ب) بررسی میزان ترانسفکشن سلولهای K562 به وسیله وکتور pEGFP-C1 زیر میکروسکوپ فلئورسنت. الف) تصویر سلولهای K562 ترانسفکت شده در نور فلورسانس ب) تصویر سلولهای ترانسفکت شده K562 در نور معمولی.(تصاویر فلورسانس در طول موج 490 نانومتر و دو روز بعد از الکتروپوریشن تهیه شده است. بازدهی الکتروپوریشن در ولتاژ 269 ولت و ظرفیت خازنی 1050 میکروفاراد 25% میباشد).[WU1]
بخش سلولی: ترانسفکشن سلولهای K562به روش الکتروپوریشن: به منظور بهینهسازی پارامترهای الکتروپوریشن سلولهای K562 ، پلاسمید pEGFP-C1 تحت شرایط مختلف به درون سلولها الکتروپوریت شد(شکل 5 الف). دو روز بعد از ترانسفکشن، سلولها زیر میکروسکوپ فلئورسنت مشاهده و میزان ترانسفکت شدن آنها بررسی شد. بهترین کارآیی الکتروپوریشن در ولتاژ 269 ولت و ظرفیت خازنی 1050 میکروفاراد به دست آمد که در این حالت درصد سلولهای ترانسفکت شده حدود 25% بود (شکل 5 ب). تعیین حداقل دوز کشندگی پرومایسین: با توجه به این که نشانگر انتخابی روی وکتور ساخته شده، مقاوم به پرومایسین بود، ابتدا کمترین غلظت از آنتیبیوتیک که سلولهای K562 غیرترانسفکت را ظرف دو هفته از بین میبرد، تعیین شد. بر این اساس پرومایسین در غلظتهای 0 تا 6 میکروگرم در هر میلیلیتر محیط کشت هر یک روز در میان به محیط کشت سلول اضافه شد و در هر بار تعویض محیط کشت، زندهمانی سلولها با رنگآمیزی تریپانبلو بررسی گردید. پس از مدت دو هفته، غلظت µg/mL 1 به عنوان حداقل غلظت کشندگی پرومایسین طی دو هفته تعیین شد.
شکل 6: نتیجه RT-PCR ژن Cas9.چاهک1: نشانگر وزن مولکولی 100 جفت بازی، چاهک2: تکثیر رونوشت Cas9 از نمونه سلول K562 ترانسفکت شده، چاهک3: نمونه سلول K562ترانسفکت نشده، چاهک4: محصول PCR بدون اضافه کردن cDNA (کنترل منفی)، چاهک 5 : نمونه RTminus (بررسی عدم وجود آلودگی DNA) از سلول ترانسفکت شده. چاهک 6: نشانگر وزن مولکولی 100 جفت بازی، چاهک 7 : تکثیر قطعه B2m از نمونه سلول K562 ترانسفکت شده. چاهک 8: تکثیر قطعه B2m از نمونه سلول K562ترانسفکت نشده، چاهک 9: محصول PCR بدون cDNA الگو (کنترل منفی)
شکل 7 : نمایش منحنیهای تکثیر و ذوب در واکنش Real-time PCR. (الف) منحنیهای تکثیر رونوشت ژنهای B2M وکلونهای بیانکننده Cas9 ، (ب) منحنیهای ذوب رونوشت ژنهای B2M وCas9. (ج) مقایسه بیان ژن Cas9 در 5 کلون مختلف جدا شده سلول K562-Cas9 . کلونهای آنالیز شده از لحاظ میزان بیان Cas9 به سه گروه با بیان کم(4 و 5)، متوسط(1 و 2) و بالا(3) تقسیم شدند. a ، b و c معنادار بودن آماری دادهها را نشان میدهند(0001/0 p < ).
بررسی بیان ژن Cas9 با RT-PCR : در ادامه، RT-PCR با استفاده از آغازگرهای اختصاصی ژن Cas9 روی RNA استخراج شده از سلولهای K562 ترانسفکت شده و غیرترانسفکت انجام شد. برای اطمینان از صحت کار در هنگام ساخت cDNA و نیز PCR، آزمایش RT-PCR بر روی ژن خانهدار B2M نیز همزمان انجام شد، قطعه مربوط به ژن B2M از سلولهای ترانسفکت شده و غیرترانسفکت تکثیر شد. اما تکثیر قطعه متعلق به ژن Cas9 تنها از سلولهای ترانسفکت اتفاق افتاد که حکایت از بیان این ژن در این سلولها دارد (شکل 6).
مقایسه میزان بیان Cas9 بین کلونهای مختلف سلول K562-Cas9 : کیفیت تکثیر ژنهای B2M و Cas9 با بررسی منحنیهای تکثیر و ذوب ژنهای مورد نظر بررسی شد (شکل 7). اختلاف چرخه آستانه(Ct) بین ژن هدف (Cas9) و رفرانس (B2M) به صورت ∆Ct=CtTarget - CtReference محاسبه شد. از آن جایی که میزان ∆Ct با میزان بیان ژن هدف نسبت عکس دارد، مقادیر پایینتر ∆Ct نشان از بیان بالاتر ژن هدف داشت(10). در این میان، 5 کلون آنالیز شده از سلولهای K562-Cas9 از لحاظ میزان بیان Cas9 به سه گروه با بیان کم(کلونهای 4 و 5)، متوسط(کلونهای 1 و 2) و بالا(کلون 3) تقسیم شدند. در این میان به نظر میرسد، با توجه به این که میزان بیان Cas9 در کلونیهای 4 و 5 و نیز 1 و 2 دو به دو با هم تقریباً برابر است، این احتمال وجود دارد که کلونها از سلول اولیه یکسانی مشتق شده باشند.
بحث امروزه با پیشرفت و گسترش ابزارهای مهندسی ژنتیک، یافتن راههای درمانی برای بیماریهای انسان با زمینه جهشهای ژنتیکی قابل دستیابی شده است. سیستم CRISPR/Cas9 از معروفترین ابزارهای اصلاح ژنی برای جهشهای مورد هدف میباشد. ساده، دقیق و ارزانتر بودن این روش اصلاح ژنی آن را بر سایر ابزارهای ویرایش ژنی از جمله ZFN و TALEN برتری داده است. البته شایان ذکر است که این روش نیز دارای جنبهها و آثار نامطلوب و چالشهایی در انتقال وکتورها به داخل سلول است که باید مورد توجه قرار بگیرد(11). از جمله این چالشها میتوان به اندازه وکتورهای مهندسی شده، تعداد وکتورها، روش انتقال آنها و نوع سلول میزبان (چسبنده یا معلق بودن آنها) اشاره نمود(12). در این مطالعه برای رفع این مشکل تصمیم گرفتیم تا ژن Cas9 را در سلولهای هدف که در اینجا سلولهای رده خونی K562 بودند، وارد کنیم تا در مطالعههای بعدی دیگر نیازی به وجود این ژن بر روی سازه ژنی نباشد. در پلاسمیدهای حامل اجزای CRISPR، بخش عمدهای از اندازه پلاسمید توسط کاست ژنی رمزکننده Cas9 (حدود 5 کیلو جفت باز) اشغال میشود. مزیت دیگری که تولید این چنین رده سلولی دارد این است که مشکل انتقال چندین وکتور به طور همزمان را حل میکند زیرا این چالش کارآیی این سیستم را کاهش میدهد(13، 12). در صورتی که سلولی ژن Cas9 را در خود بیان کند، مانند آن چه که در این مطالعه صورت گرفته، دیگر نیازی به وجود آن بر روی پلاسمید نیست و تنها وجود کاست رمزکننده sgRNA به همراه توالیهای پروکاریوتی روی وکتور کفایت میکند. لازمه ساخت رده سلولی K562 بیانکننده Cas9، ساخت وکتوری بود که از طرفی حامل کاست بیانی Cas9 بوده و از سوی دیگر دارای یک نشانگر انتخابی که در اینجا ژن مقاومت به پرومایسین بود، باشد. با توجه به این که چنین پلاسمیدی در داخل در دسترس نبود، ابتدا ساخت آن در دستور کار قرار گرفت. بدین منظور، از وکتور pCas-Guide-AAVS1 که حامل کاست بیانی Cas9 بود به عنوان وکتور مبنا استفاده شد. با بررسی این وکتور مشخص گردید که هضم آنزیمی آن با دو آندونوکلئاز KpnI و EcoRI امکان وارد کردن کاست بیانی مقاومت به پرومایسین را فراهم میکند. کاست ژن مقاومت به آنتیبیوتیک نیز با PCR از وکتور pAAVS1-puro-DNR با تعبیه دو جایگاه برشی مدنظر در طرفین آن تکثیر شد. همسانهسازی قطعه PCR در وکتور مبنا منجر به ساخت پلاسمیدی به نام pPPC-Cas به طول 9 کیلو جفت باز شد. از دیگر چالشهای این مطالعه میتوان به سخت بودن ترانسفکت کردن سلولهای K562 اشاره نمود. مطالعههای گذشته حاکی از آن است که به طور کلی ترانسفکت کردن سلولهای معلق کار دشوارتری نسبت به ترانسفکت سلولهای چسبنده میباشد(14). در این مطالعه از روش الکتروپوریشن برای ترانسفکت کردن سلولهای K562 استفاده گردید. میزان مرگ و میر در این روش بالا است(13). اما از طرفی با استفاده از این روش میتوان در مدت زمان کوتاهی تعداد زیادی از سلولها را با سمیت کم به سادگی ترانسفکت کرد(15، 14). برای بهینه کردن شرایط الکتروپوریشن سلولهای K562 از وکتور pEGFP-C1 که حامل EGFP است، استفاده شد. وجود ژن EGFP باعث شد تا درصد سلولهای ترانسفکت شده به وسیله میکروسکپ فلئورسنت قابل پیگیری و تعیین باشد. الکتروپوریشن به صورت تصاعدی در ولتاژها و ظرفیتهای خازنی متفاوت بررسی شد که در نهایت بهترین شرایط الکتروپوریشن به صورت 269 ولت وµF 1050(میکروفاراد) انتخاب گردید که در آن حدود 25% از سلولها ترانسفکت میشدند. در مرحله بعد، برای ساخت سلولهای K562 بیانکننده Cas9 وکتور ساخته شده در این پژوهش به درون سلولها با شرایط بهینه شده الکتروپوریشن منتقل شد. با توجه به وجود ژن مقاومت به پرومایسین در وکتور ساخته شده، سلولهای دریافتکننده پلاسمید با این آنتیبیوتیک در غلظت µg/mL 1 جدا شدند. زیرا قبلاً حداقل دوز کشندگی پرومایسین که سلولهای K562 غیرترانسفکت را طی دو هفته از بین میبرد به میزانg/mL 1 تعیین شده بود. انتخاب سلولها با پرومایسین دو هفته به طول انجامید و این مدت زمان برای این در نظر گرفته شد تا تنها سلولهایی جدا شوند که ژن مقاومت به آنتیبیوتیک وارد ژنوم آنها شده است و پلاسمید را به صورت اپیزومال در خود حمل نمیکنند. به علت عدم وجود منشا همانندسازی پستاندار در وکتور ساخته شده pPPC-Cas، در صورتی که پلاسمید به داخل ژنوم سلول میزبان وارد نشود، طی دو هفته تقسیم و تکثیر سلولی، پلاسمیدهای اپی زومی از بین رفته و تنها سلولهایی که به صورت پایدار ترانسفکت شدهاند، قادر به رشد در محیط انتخابی(حاوی پرومایسین) خواهند بود. ورود وکتور pPPC-Cas به داخل ژنوم رده سلولی K562 بعد از الکتروپوریشن به صورت تصادفی میباشد. این نوع ورود به ژنوم میتواند مشکلاتی را در پی داشته باشد. اول این که ورود تصادفی ممکن است منجر به تخریب کاست بیانی Cas9 گردد. چرا که در این حالت وکتور میتواند از هر بخش خود به صورت شانسی نوترکیبی داده و وارد ژنوم شود. بنابراین با توجه به این که بخش عمدهای از وکتور ساخته شده در برگیرنده کاست بیانی Cas9 است، این امر محتمل میباشد که وارد شدن به داخل ژنوم به کاست ژن Cas9 آسیب وارد کند. مسئله دوم این که ورود تصادفی پلاسمید به داخل ژنوم میتواند باعث شود تا میزان بیان ژن Cas9 در سلولهای مختلف متفاوت باشد. حتی ممکن است در برخی از سلولها، بیان ژن Cas9 خاموش شود. دلیل این موضوع آن است که محل ورود ژن به داخل ژنوم میتواند روی بیان آن تاثیرگذار باشد که به آن اثر جایگاه کروموزومی (Chromosomal position effect) میگویند. تمامی نقاط ژنوم به لحاظ فعالیت رونوشت برداری یکسان نیستند. برای مثال ژن ممکن است وارد یک نقطهای از DNA ژنومی شود که در آن رونویسی از ژنها بسیار فعال است. این موضوع میتواند باعث کاهش بیان و حتی خاموش شدن بیان ژن انتقال یافته شود و مشکل آخر این که در ورود تصادفی، تعداد نسخههای ژن وارد شده به داخل ژنوم قابل کنترل نیست و تعداد نسخههای ژن بین سلولهای مختلف متفاوت میباشد. تعدد ورود ترانس ژن به داخل ژنوم اثر قابل پیشبینی ندارد. در مواردی مشاهده شده است تعدد نسخهها میتواند اثر افزایشی روی بیان ژن هدف بگذارد، اما گاهی نیز اثر منفی روی بیان ژن و حتی رشد سلول گزارش شده است(16). همه این مشکلات میتواند منجر به بیان هتروژن ژن هدف در میان سلولهای ترانسفکت شود. بنابراین برای رفع این مشکلات انتخاب کلونی انجام شد. 5 کلونی از سلولهای K562 مقاوم به پرومایسین انتخاب شده و پس از رشد و تکثیر، RNA آنها به طور جداگانه استخراج و بیان ژن Cas9 در آنها با Real-time PCR بررسی شد. برای اطمینان از سالم بودن کاست بیانی Cas9 و این که رونوشتهای کامل و سالم از ژن Cas9 وجود دارد، آغازگرها به گونهای طراحی شدند که انتهای ´3 رونوشت Cas9 را شناسایی و تکثیر میکردند. نتیجه آنالیز بیان Cas9 در 5 کلونی جدا شده نشان داد که بیان Cas9 در بین آنها متفاوت است. از میان 5 کلونی آنالیز شده به نظر میرسید منشا برخی از کلونها یکسان میباشد و در واقع از سلول ترانسفکت شده مشترک مشتق شدهاند.
نتیجهگیری نتیجه تحقیق حاضر تولید سلول K562-Cas9 است که
ژن Cas9 را به صورت دائم بیان میکند. سلولهای K562 سلولهای اریترولوکمیای انسانی هستند که در تحقیقات بیماریهای خونی از جمله هموگلوبینوپاتیها مانند β-تالاسمی بسیار استفاده میشوند. لذا دست آورد این پژوهش میتواند ابزاری برای کمک به این دسته از پژوهشها در کشور باشد. از سویی دیگر تجربه حاصل از این کار میتواند در توسعه سایر ردههای سلولی بیانکننده Cas9 نیز مورد استفاده قرار گیرد.
تشکر و قدردانی این مقاله منتج از پایاننامه دانشجویی کارشناسی ارشد مؤسسه عالی آموزشی و پژوهشی طب انتقال خون میباشد. مراتب قدردانی خود را از تمام کسانی که در این کار پژوهشی همکاری نمودهاند به ویژه پژوهشگاه ملی مهندسی ژنتیک و زیست فنآوری اعلام میدارم.
Ensari F, Nikougoftar M, Hamidieh A, Shamsara M. Generation of K562 cell line expressing Cas9 endonuclease (CRISPR-associated9). Sci J Iran Blood Transfus Organ 2019; 16 (1) :32-43 URL: http://bloodjournal.ir/article-1-1223-fa.html
انصاری فائزه، نیکوگفتار ظریف مهین، حمیدیه امیرعلی، شمس آرا مهدی. تولید رده سلولی K562 بیان کننده اندونوکلئاز Cas9 (CRISPR-associated9). فصلنامه پژوهشی خون. 1398; 16 (1) :32-43